|
Glob Reg Health Technol Assess 2025; 12: 43-48 ISSN 2283-5733 | DOI: 10.33393/grhta.2025.3469 POINT OF VIEW |
L’assessment e il valore del farmaco: una sintesi dei focus group della XXII Conferenza Nazionale sulla Farmaceutica
Assessment and value of drugs: report of the focus groups from the XXII National Conference on Pharmaceuticals
The XXII National Conference on Pharmaceuticals, held in Catania from 29 February to 1 March 2024, involved the participation of representatives from more than forty pharmaceutical industries, national authorities, academia, clinicians and clinical pharmacists. The 22nd edition represented a key forum for the analysis of value assessment of medicines, focusing on the impact of new European and National regulations, including the Italian Medicines Agency (AIFA) reform and the HTA regulation. This point of view summarizes insights from focus groups discussions that took place during the Conference, highlighting the pillar role of the new HTA regulation for homogenizing the evaluation across Europe, and the importance to strengthen the collaboration among the parties involved from an early phase, and implementing adaptive and flexible assessment, especially for orphan medicines. The re-evaluation of the innovativeness criteria and framework of the Pricing and Reimbursement (P&R) dossier are points raised among the different focus groups, especially for aligning these tools to the upcoming HTA regulation. The proposals emerged could be useful for AIFA, which is facing a reorganization aimed to optimizing the reimbursement process in Italy.
Keywords: Access, Health Technology Assessment, HTA regulation, Innovativeness, Orphan drugs
Received: January 17, 2025
Accepted: February 15, 2025
Published online: February 25, 2025
Global & Regional Health Technology Assessment - ISSN 2283-5733 - www.aboutscience.eu/grhta
© 2025 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0).
Commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Introduzione
La XXII Conferenza Nazionale sulla Farmaceutica, tenutasi a Catania dal 29 febbraio al 1° marzo 2024, ha rappresentato un forum importante per l’analisi della valutazione del valore dei medicinali, concentrandosi sull’impatto delle nuove Normative Europee e Nazionali, tra cui la riforma dell’Agenzia Italiana del Farmaco (AIFA) emanata con decreto ministeriale dell’8 gennaio 2024 no 3 e pubblicata nella Gazzetta Ufficiale della Repubblica Italiana (GURI) il 15 gennaio 2024 (1) e il nuovo Regolamento (UE) 2021/2282 sull’Health Technology Assessment (HTA) (2).
Oltre alla sessione plenaria, che ha visto la partecipazione di Accademia, AIFA, Società Scientifiche e Industria, la Conferenza prevedeva anche un momento di confronto su specifiche tematiche di elevato interesse, attraverso la partecipazione a focus group. Pur affrontando tematiche diverse, si è seguito un format analogo che prevedeva l’identificazione dello stato dell’arte, delle criticità e delle proposte. Ogni focus group era guidato da una moderatrice, proveniente da una delle aziende partecipanti, supportata nella gestione della discussione, raccolta delle criticità e delle proposte emerse da un facilitatore presente in aula e da un medical writer per la redazione finale della presentazione e del documento di sintesi. Inoltre, durante la conferenza le moderatrici hanno avuto anche l’occasione di esporre quanto emerso.
Ai focus group hanno preso parte più di 70 partecipanti provenienti da 30 aziende farmaceutiche e professionisti provenienti dal mondo accademico; ad ogni partecipante è stata data la possibilità di scegliere a quale focus group partecipare, tra i quattro che riguardavano tematiche diverse:
- aggiornamenti dei criteri di innovatività;
- criticità regolatorie nel disegno dei trial clinici;
- farmaci per malattie rare e orfane;
- il nuovo Regolamento HTA.
Le criticità e le proposte emerse che vengono di seguito sintetizzate hanno evidenziato che, pur affrontando tematiche diverse, in realtà vi è un fil rouge che passa attraverso queste quattro dimensioni strettamente interconnesse.
Focus group: Aggiornamento dei criteri di innovatività
La discussione all’interno di questo focus group ha evidenziato la necessità di ripensare agli attuali criteri per l’attribuzione dell’innovatività (3), considerando il contesto che affronta una fase di importante cambiamento.
Pertanto, sarà importante comprendere l’interdipendenza di tali cambiamenti e come impatteranno sugli attuali criteri per la classificazione dei farmaci innovativi.
Il nuovo Regolamento europeo HTA (2) ha tra i suoi obiettivi la definizione di misure per la valutazione clinica congiunta delle tecnologie sanitarie, il Joint Clinical Assessment (JCA), la scelta dei comparatori sulla base delle alternative terapeutiche a livello europeo e la definizione degli outcome di interesse.
Sarà quindi di fondamentale importanza comprendere quale ruolo avrà l’AIFA nella definizione dei PICO (Patient, Intervention, Comparison, Outcome) e inoltre come verrà utilizzato il JCA nelle stesse valutazioni dell’innovatività.
Partendo da queste considerazioni, si è proceduto con un’analisi critica degli attuali criteri di valutazione dell’innovatività per proporre possibili soluzioni per il futuro, evidenziando a oggi che:
- il coinvolgimento delle Società Scientifiche e delle Associazioni di Pazienti è limitato e non strutturato;
- le rivalutazioni dei farmaci con innovatività condizionata sono limitate;
- il Fondo per i farmaci innovativi non è appropriatamente utilizzato.
Analizzando singolarmente i tre domini (bisogno terapeutico, valore terapeutico aggiunto e qualità delle prove) (4) emerge come il valore terapeutico aggiunto sia il principale driver di valutazione dell’innovatività (5-7); inoltre è emerso che la definizione del bisogno terapeutico ha oggi un peso meno rilevante nel giudizio di innovatività e probabilmente richiederebbe il coinvolgimento delle Associazioni di Pazienti per definire il peso che realmente avrebbe l’introduzione della terapia (8-11).
Tuttavia, manca una definizione condivisa di cosa sia “clinicamente rilevante” a seconda dell’area terapeutica e gli endpoint considerati sono poco flessibili e quindi poco adattabili alle differenti aree terapeutiche o al setting di malattia. Inoltre, anche i criteri per la definizione dei comparatori sono un punto critico e sono correlati anche al ruolo che potrebbero avere le comparazioni indirette e i dati di Real-World Evidence (RWE) a supporto degli studi pivotali (12-14).
Per quanto riguarda la qualità delle prove, la metodologia GRADE (Grading of Recommendations, Assessment, Development and Evaluation) può presentare dei limiti poco adattabili e trasversali alle diverse aree terapeutiche (15).
Tra le possibili soluzioni trasversali, si annoverano la necessità di scoping meeting tra AIFA e Industria come momenti propedeutici alla definizione dei PICO e la ridefinizione del ruolo contestuale di AIFA in modo che abbia più peso nella determinazione dei PICO stessi e tutto ciò al fine di integrare al meglio il JCA nel processo di definizione dell’innovatività.
Anche la gestione delle tempistiche di accesso dei farmaci dovrebbe subire un’evoluzione al fine di garantire l’inclusione nei prontuari in modo veloce ed equo in tutte le Regioni (16, 17); a tale scopo le proposte discusse hanno riguardato la possibilità di istituire una gara unica a livello nazionale che possa portare a un’armonizzazione in questo contesto. Per quanto riguarda poi i due gradi di innovatività, è emerso che si potrebbe concedere fin da subito l’accesso al Fondo anche ai farmaci con innovatività potenziale, definendo una deadline che permetta la raccolta e la presentazione di ulteriori studi o dati a sostegno del mantenimento dell’innovatività.
Sulla base di quanto emerso, si riportano di seguito le soluzioni proposte per un aggiornamento dei criteri dell’ innovatività (Fig. 1).
Focus group: Criticità regolatorie nel disegno dei trial clinici
Questo focus group ha approfondito le criticità relative al disegno degli studi clinici rispetto al ruolo nella definizione della strategia di accesso.
I partecipanti hanno riconosciuto che i disegni di sperimentazione tradizionali spesso catturano in modo inadeguato la complessità nella gestione terapeutica e hanno pertanto valutato quanto sia importante introdurre metodologie di sperimentazione innovative che sfruttino dati di RWE e disegni adattativi per migliorare la partecipazione, garantendo comunque solide valutazioni di sicurezza e di efficacia (12-14).
Guardando al ruolo delle Autorità regolatorie nella valutazione del profilo clinico di un farmaco sulla base delle evidenze presentate, è stato evidenziato che vi è un disallineamento tra gli obiettivi di valutazione tra EMA, AIFA e gli organismi regionali. È stata evidenziata la necessità di armonizzare i requisiti per dimostrare benefici e rischi, nonché gli endpoint, al fine di facilitare la valutazione e l’accesso ai trattamenti.
Infatti, le differenze in termini di obiettivi valutativi tra EMA e le singole Agenzie regolatorie nazionali si traducono in criticità e nel disallineamento tra i singoli Stati Membri. Se la mission dell’EMA è quella di rendere disponibile e di regolamentare l’accesso del farmaco basandosi su criteri di efficacia e sicurezza clinica “assoluti”, le singole Agenzie Nazionali (come AIFA) possiedono uno scopo diverso, vale a dire garantire anche la sostenibilità del Sistema Sanitario, quindi una valutazione “relativa” del medicinale (rapporto costo/beneficio rispetto alle alternative disponibili). Risulta evidente quindi che, nella definizione dei trial clinici e nel loro disegno, è necessario confrontarsi con questa differenza di obiettivi tra EMA e le singole Agenzie Nazionali. In aggiunta, talvolta, le decisioni dell’EMA vengono messe in discussione dalle singole Agenzie e/o dalle singole aziende, in termini di definizione del place in therapy del farmaco, che spesso viene condizionato (ristretto) da limiti economici derivanti dalla negoziazione (18).
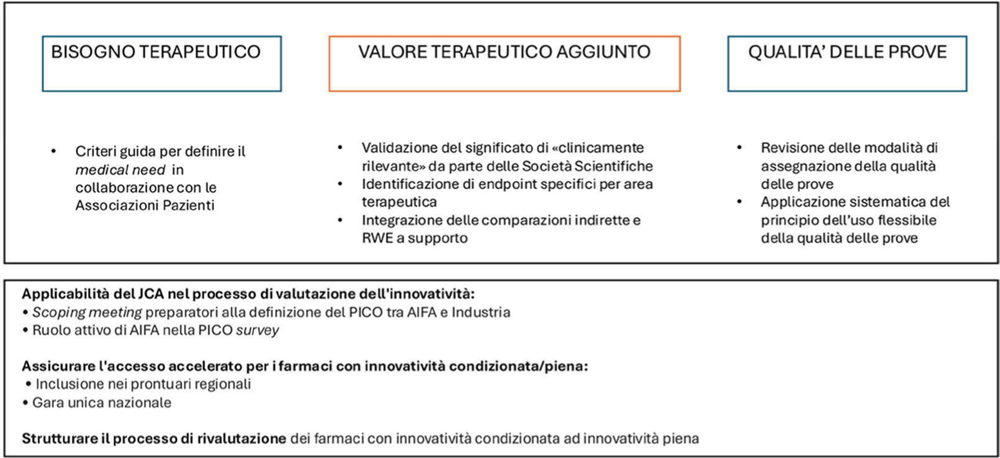
FIGURE 1 - Proposte del focus group sull'aggiornamento dei criteri di innovatività.
È stata evidenziata l’importanza di rafforzare la cooperazione tra enti regolatori, industria e Associazioni di Pazienti, anche in questo contesto; la collaborazione tra queste parti è vitale per sviluppare progetti di studi clinici su misura, soprattutto se si affrontano le sfide uniche associate a popolazioni di pazienti affetti da patologie rare (19, 20).
Il tema del conflitto di interessi è risultato critico e complesso, perché spesso, infatti, la figura esperta che si occupa della valutazione del farmaco e dello studio ad esso associato è quella che inevitabilmente possiede un conflitto di interessi. Per l’Agenzia potrebbe infatti risultare complicato trovare dei clinici esperti per la valutazione del farmaco che siano allo stesso tempo privi di conflitti di interessi. Dal gruppo di lavoro è emerso come esista una rigidità assoluta in termini di “conflitto di interessi”, che quindi risulta essere difficile da gestire in quanto tutti gli stakeholder sono caratterizzati dal conflitto di interessi, paziente compreso.
Il punto chiave più volte emerso è la necessità di cooperare per uno scopo comune, attraverso il rafforzamento di trasparenza e dialogo costruttivo tra AIFA e aziende, che dovrebbe iniziare in fase pre-submission e mantenersi durante il ciclo di vita del farmaco, permettendo per esempio la condivisione da parte dell’AIFA dei documenti istruttori. È stato proposto di stabilire un early dialogue e uno scoping meeting tra aziende e Commissione Scientifica ed Economica del Farmaco (CSE) di AIFA. Accanto alla cooperazione basata sul dialogo, si dovrebbero sfruttare maggiormente i dati real-life, inclusi i dati provenienti dai registri di monitoraggio, che rappresentano una fonte preziosa di dati, soprattutto in un’ottica di rivalutazione del farmaco o in presenza di nuove indicazioni (Fig. 2).
Focus group: Farmaci per malattie rare e orfane
Questo focus group si è concentrato sulle sfide specifiche legate ai medicinali per le malattie rare e orfane di terapia e dei relativi quadri normativi. I partecipanti hanno convenuto che le normative attuali non supportano in modo adeguato i requisiti specifici di queste terapie, rendendo necessario uno spostamento verso approcci più flessibili e adattativi in grado di semplificare il processo di approvazione e di migliorare l’accesso.

FIGURE 2 - Proposte del focus group sulle criticità regolatorie dei trial clinici.
Inoltre, sono state evidenziate le difficoltà specifiche legate all’accesso alle cure per le malattie rare, come i tempi lunghi per la loro immissione in commercio in Italia, in quanto, dopo l’approvazione da parte dell’EMA, devono superare sia lo “scoglio” della rimborsabilità a livello nazionale, regionale e locale sia la mancanza di strumenti efficaci di accesso precoce. Alla luce di ciò, vi è l’urgente necessità di esplorare schemi alternativi di regolamentazione per la commercializzazione e il rimborso, che possano risolvere efficacemente le disparità esistenti in termini di accesso, promuovendo al tempo stesso un’innovazione coerente con le sfide uniche presentate dalle malattie rare, che comportano paradigmi terapeutici complessi e popolazioni limitate di pazienti.
Partendo dal place in therapy dei farmaci orfani, la valutazione dell’EMA non dovrebbe differire da quella definita a livello nazionale, come evidenziato anche nel gruppo delle criticità del disegno degli studi clinici (18). Si è discusso sulla necessità di concordare dei criteri flessibili nella valutazione degli endpoint e di includere anche le comparazioni indirette tra gli strumenti di scelta dei comparatori, quando non vi è un’alternativa terapeutica.
Risulta fondamentale la corretta applicazione della procedura di fast track, rispettando la tempistica dei 100 giorni (21), prioritizzando le procedure per i farmaci per malattie orfane e rare. Sarebbe auspicabile creare uno specifico strumento legislativo di early access dedicato ai farmaci destinati al trattamento di malattie orfane e rare, rifacendosi a modelli europei, pur adattati al contesto italiano, e istituire scoping meeting con gli uffici AIFA nella fase pre-submission del dossier.
È stata inoltre sottolineata la necessità di rivedere i criteri di valutazione dell’innovatività (4) da applicare a questi farmaci, rivalutando i valori assegnati all’unmet need e al valore terapeutico aggiunto e dando maggiore peso alle evidenze disponibili, anche se limitate.
Guardando all’attuale struttura del dossier Prezzo & Rimborso (P&R) (22), questo dovrebbe adattarsi nel caso di farmaci orfani e dare la possibilità di inserire dati epidemiologici e proxy in presenza di malattie ultra-rare; dovrebbero anche essere identificati criteri specifici per le valutazioni economiche da applicare ai farmaci orfani.
I partecipanti hanno evidenziato che vi è un’inadeguata attenzione nei confronti dei prodotti antibiotici “reserve” utilizzati come ultima linea di trattamento nei casi di infezioni multi-resistenti.
Spostando l’obiettivo all’accesso a livello regionale si dovrebbero creare dei percorsi di horizon scanning per permettere un dialogo precoce tra AIFA e Regioni al fine di definire direttive per l’accesso. Inoltre, al fine di velocizzare l’accesso regionale, il gruppo di lavoro propone:
- l’abolizione di PTR e PTOR;
- la valutazione del setting di cura e del medico prescrittore tramite un unico decreto dirigenziale;
- l’immediatezza delle procedure di acquisto, anche tramite una procedura negoziata o acquisti sottosoglia (per esempio, affidamenti diretti tramite ASL o Aziende Ospedaliere), nelle more dell’espletamento della gara regionale.
Ulteriore e fondamentale aspetto a cui prestare attenzione è la presa in carico olistica del paziente con malattia rara, senza differenze da Regione a Regione, tramite l’aggiornamento dei Livelli Essenziali di Assistenza (LEA) e la presa in carico degli Extra LEA, fin dalla fase della diagnosi, e l’implementazione di Percorsi diagnostici terapeutico-assistenziali (PDTA) in modo uniforme su tutto il territorio nazionale.
Un approccio più flessibile nonché una collaborazione rafforzata tra le parti interessate potrebbero facilitare il superamento di tortuosità burocratiche e garantire un accesso tempestivo ai trattamenti essenziali per i pazienti affetti da malattie rare. In questo contesto è emerso il ruolo chiave dei partenariati pubblico-privato, che potrebbero snellire il percorso dell’accesso di nuovi strumenti terapeutici (Fig. 3).

FIGURE 3 - Proposte del focus group sui farmaci per malattie rare e orfane.
Focus group: Il nuovo Regolamento HTA
Il focus group sull’HTA ha esaminato attentamente le implicazioni del previsto Regolamento europeo HTA (2), la cui applicazione è avvenuta il 12 gennaio 2025.
Lo scopo precipuo di questo Regolamento è quello di uniformare le procedure di valutazione delle tecnologie sanitarie tra i diversi Stati Membri, promuovendo così un processo decisionale armonizzato in merito all’accesso dei medicinali oggetto dell’assessment.
Tra i potenziali benefici derivanti dall’armonizzazione delle valutazioni HTA a livello europeo, vi sono la riduzione della duplicazione delle valutazioni sul beneficio terapeutico e il miglioramento dell’accessibilità. Tuttavia, i partecipanti hanno sottolineato l’esigenza di preservare un quadro di valutazione flessibile e reattivo sulla base delle caratteristiche distintive delle terapie, anche guardando allo specifico contesto nazionale.
Nonostante gli obiettivi di armonizzazione del Regolamento, sono state espresse perplessità circa la sua l’implementazione pratica e su quanto si è realmente pronti ad affrontare il cambiamento, sia a livello delle Autorità regolatorie quanto a livello aziendale (23).
I partecipanti hanno sottolineato quanto sia importante dover puntare alla formazione e alla necessità di strutturare nuove figure aziendali specializzate che possano supportare le attività richieste dal Regolamento.
Esiste la convinzione che il successo del nuovo quadro normativo sull’HTA dipenderà in larga misura dalla promozione di una collaborazione sinergica tra le diverse parti interessate, proponendo anche la possibilità di istituire un gruppo di lavoro sull’implementazione della regolamentazione dell’HTA, che debba riunire rappresentanti del Ministero della Salute, AIFA, accademia, aziende, cittadini, pazienti e Società Scientifiche, con l’obiettivo comune di porre l’Italia come attore principale a livello europeo.
È stata espressa all’unanimità la necessità di coinvolgere l’accademia, in grado di supportare la formazione degli operatori del settore, anche per affrontare al meglio il tema dei PICO.
Alla base del successo dell’applicazione del Regolamento vi è la collaborazione tra tutte le parti, che dovrebbe basarsi sull’early dialogue per produrre input e condividere conoscenze utili per i PICO e le dimensioni non cliniche; si dovrebbe inoltre ripensare alla struttura del dossier P&R (22), che deve essere coerente con il nuovo schema JCA, e bisognerebbe adattare i criteri di innovatività (4) al nuovo contesto, così come ripensare ai MEA (Managed Entry Agreement) e alla metodologia per l’integrazione tra JCA e procedura di rimborso.
I partecipanti hanno inoltre sottolineato l’importanza di garantire l’indipendenza del processo di valutazione delle tecnologie sanitarie da influenze esterne, al fine di preservare la credibilità e la legittimità delle valutazioni prodotte. Questa indipendenza è fondamentale per alimentare la fiducia nei risultati dell’HTA, poiché la percezione di eventuali pregiudizi potrebbe minare la fiducia del pubblico nel processo di valutazione e, di conseguenza, avere ripercussioni sull’accesso dei pazienti alle terapie. Ciò evidenzia la necessità di istituire solide strutture di governance che diano priorità alla trasparenza e alla responsabilità nelle decisioni relative all’HTA (Fig. 4).

FIGURE 4 - Proposte del focus group sul Regolamento HTA.
Conclusioni
Nonostante la diversità dei temi affrontati, le proposte emerse dai confronti instaurati all’interno dei focus group presentano dei fattori comuni, a rimarcare il filo conduttore che esiste dallo sviluppo clinico all’accesso sul mercato.
La revisione dei criteri di innovatività è un tema più volte affrontato, sia nel contesto dei farmaci orfani sia in relazione al nuovo Regolamento HTA, così come più volte è stata rimarcata la necessità di rivalutare alcuni aspetti del dossier P&R, che possa adattarsi al JCA anche in presenza di malattie rare e ultra-rare.
Comune denominatore emerso è la necessità di instaurare approcci collaborativi e trasparenti allo scopo di incorporare le prospettive delle diverse parti interessate, rafforzando l’early dialogue e lo scoping meeting, a prescindere dalla tecnologia sanitaria da affrontare. In questo contesto è emerso il ruolo dell’accademia, che non dovrebbe essere marginale ma centrale per accrescere conoscenza e competenza.
Favorire un contesto collaborativo tra le diverse parti interessate, dalle Autorità regolatorie alle organizzazioni dei pazienti, sarà essenziale per sviluppare un quadro normativo per l’HTA che non solo sia solido ed efficace ma che rifletta le complesse realtà in cui le terapie saranno implementate.
Acknowledgements
We wish to thank all the focus groups’ participants who contributed to the discussion and proposals outlined in this work:
Giovanni Alquati (Gilead Sciences), Sonia Amore (Novartis), Sabrina Baldanzi (Grunenthal), Michele Basile (Altems Advisory), Federica Basso (Advanz Pharma), Silvi Beghin (Menarini Ricerche), Fabio Blandino (Ipsen), Filippo Bova (Mundipharma), Filippo Bova (Mundipharma), Filippo Bresciani (Ipsen), Pietro Brambilla (Lundbeck), Elisabetta Brioschi (Daiichi-Sankyo), Stefania Mazzocchi (Advanz Pharma), Sergio Cipolla (GSK), Fernando Crisarà (MSD), Elisa Crovato (Chiesi), Andrea Giuseppe Curia (SOBI), Lorenzo Cultrona (Lundbeck), Lucia D’Angiolella (Novo Nordisk), Eugenio Di Brino (Altems Advisory), Gianluca Di Dario (Novartis), Nello Donnini (AbbVie), Francesco Fatiga (Janssen), Matteo Ferrario (Roche), Fausto Galanti (Bayer), Armando Genazzani (Prof. Ordinario Farmacologia, Torino), Giampiero Geusa (CSL Vifor), Rosalba Gregorini (Gilead Sciences), Giorgia Guareschi (Novo Nordisk), Marco Guazzini (Jazz Pharmaceutical), Cristina Iaccarino (CSL Vifor), Paola Ianna (Daiichi-Sankyo), Yosra Kamel (GSK), Nicoletta Martone (SOBI), Elisa Martelli (Gilead Sciences), Isabella Masserini (Grunenthal), Gioele Maverna (Ipsen), Manuela Marrano (SIFI), Pier Paolo Mangia (Chiesi), Marta Meloncelli (Alexion), Mario Napoli (Menarini Ricerche), Carmelo Milazzo (Menarini Ricerche), Corrado Minelli (CSL Vifor), Cinzia Musella (Alexion), Vincenzo Papa (SIFI), Domenico Pancrazzi (Jazz Pharma), Maria Assunta Pennacchia (CSL Vifor), Leonardo Perrone (AbbVie), Andrea Pierini (Roche), Lara Pippo (CSL Behring), Iolanda Porto (SIFI), Giuseppe Pompilio (Janssen), Eleonora Premoli (Idorsia Pharmaceuticals), Emanuele Pria (Gilead Sciences), Roberto Pala (Menarini Ricerche), Filippo Rumi (Altems Advisory), Susan Sammak (Amgen), Valentina Straziota (Daiichi-Sankyo), Antonia Tricarico (Bayer), Elena Trovati (Amgen), Bruna Zanocchia (Novartis).
A special thanks to the coordinators who contributed during the conference and on the drafting of the manuscript.
Disclosures
Conflict of interest: TM is an employee of Lundbeck Italia; FP is an employee of AstraZeneca Spa; FR is an employee of Novartis Farma. VD, CT and FD declare no conflict of interest.
Financial support: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Bibliografia
- 1. AIFA. Decreto 8 gennaio 2024, n.3, Regolamento recante modifiche al regolamento sull’organizzazione e sul funzionamento dell’Agenzia Italiana del Farmaco, GU Serie Generale n. 11 del 15 gennaio 2024. (Accessed January 2025) Online
- 2. Regolamento (UE) 2021/2282 del Parlamento Europeo e del Consiglio del 15 dicembre 2021 relativa alla valutazione delle tecnologie sanitarie e che modifica la Direttiva 2011/24/UE. (Accessed January 2025) Online
- 3. Gazzetta Ufficiale della Repubblica Italiana. Determina 1535/2017 dell’Agenzia Italiana del Farmaco “criteri per la classificazione dei farmaci innovativi, e dei farmaci oncologici innovativi, ai sensi dell’articolo 1, comma 402 della legge 11 dicembre 2016, n. 232, GU n. 218 del 18 settembre 2017”. (Accessed January 2025) Online
- 4. AIFA. Modulo per la richiesta del riconoscimento dell’innovatività. (Accessed January 2025) Online
- 5. Jommi C, Galeone C. The Evaluation of Drug Innovativeness in Italy: Key Determinants and Internal Consistency. PharmacoEconom Open. 2023;7(3):373-381. CrossRef PubMed
- 6. Fortinguerra F, Perna S, Marini R, Dell’Utri A, Trapanese M, Trotta F; Scientific & Technical Committee (Commissione Tecnico-Scientifica, CTS) of Italian Medicines Agency-AIFA. The assessment of the innovativeness of a new medicine in Italy. Front Med (Lausanne). 2021;8(1):793640. CrossRef PubMed
- 7. Galeone C, Bruzzi P, Jommi C. Key drivers of innovativeness appraisal for medicines: the Italian experience after the adoption of the new ranking system. BMJ Open. 2021;11(1):e041259. CrossRef PubMed
- 8. Jommi C, Armeni P, Bertolani A, Costa F, Otto M. Il futuro dei Fondi per Farmaci Innovativi: risultati di uno studio basato su Delphi panel. Glob Reg Health Technol Assess. 2021;8:22-28. CrossRef PubMed
- 9. Lobban T CA and Camm. Patient associations as stakeholders: a valuable partner for facilitationg access to therapy. Europace. 2011, 13, ii21-ii24:10. CrossRef PubMed
- 10. Ocloo J, Matthews R. From tokenism to empowerment: progressing patient and public involvement in healthcare improvement. BMJ Qual Saf. 2016;25(8):626-632. CrossRef PubMed
- 11. Haerry D, Landgraf C, Warner K, et al. EUPATI and patients in medicines research and development: guidance for patient involvement in regulatory processes. Front Med (Lausanne). 2018;5:230. CrossRef PubMed
- 12. Graffigna G, Barello S, Riva G, et al. Italian Consensus Statement on Patient Engagement in chronic care: process and outcomes. Int J Environ Res Public Health. 2020;17(11):4167. CrossRef PubMed
- 13. Miksad RA, Abernethy AP. Harnessing the Power of Real-World Evidence(RWE): a checklist to ensure regulatory-grade data quality. Clin Pharmacol Ther. 2018;103(2):202-205. CrossRef PubMed
- 14. Beaulieu-Jones BK, Finlayson SG, Yuan W, et al. prasad V, Yu KH. Examining the use of real-World Evidence in the regulatory Process. Clin Pharmacol Ther. 2020;107(4):843-852. CrossRef PubMed
- 15. Prilla S, Groeneveld S, Pacurariu A, et al. Real-World Evidence to Support EU Regulatory Decision Making-Results From a Pilot of Regulatory Use Cases. Clin Pharmacol Ther. 2024;116(5):1188-1197. CrossRef PubMed
- 16. GRADE Welcome to the GRADE working group. From evidence to recommendations-trasparent and sensible. Online www.gradeworkinggroup.org (Accessed January 2025)
- 17. Cicchetti A, Gasbarrini A. The healthcare service in Italy: regional variability. Eur Rev Med Pharmacol Sci. 2016;20(1)(suppl):1-3. PubMed
- 18. Bortolami A, Jommi C, Bresciani F, Piccoli L, Sangiorgi E, Scroccaro G. Regional Formularies in Italy: current state and future perspectives. Glob Reg Health Technol Assess. 2024;11(1):68-74. CrossRef PubMed
- 19. Wolters S, Jansman FGA, Postma MJ. Differences in evidentiary rewuirements between European Medicines Agency and European Health Technology Asessment of Oncology drugs-Can allignment be enhanced? Value Health. 2022;25(12): 1958-1966. CrossRef PubMed
- 20. Gentilini A, Miraldo M. The role of patient organisations in research and development: evidence from rare diseases. Soc Sci Med. 2023;338:116332. CrossRef PubMed
- 21. Shakhnenko I, Husson O, Chuter D, van der Graaf W. Elements of successful patient involvement in clinical cancer trials: a review of the literature. ESMO Open. 2024;9(4):102947. CrossRef PubMed
- 22. Decreto legge 13 settembre 2012, n. 158, articolo 12, comma 5 bis. “Disposizioni urgenti per promuovere lo sviluppo del Paese mediante un più alto livello di tutela della salute”. (Accessed January 2025) Online
- 23. AIFA. Decreto 2 agosto 2019. “Criteri e modalità con cui l’Agenzia Italiana del Farmaco determina, mediante negoziazione, i prezzi dei farmaci rimborsati dal Servizio sanitario nazionale. GU Serie Generale, n. 185 del 24 luglio 2020”. (Accessed January 2025) Online
- 24. Patarnello F. Regolamento HTA: come si sta muovendo l’Italia? Glob Reg Health Technol Assess. 2024;11:51-54. CrossRef PubMed