|
Glob Reg Health Technol Assess 2024; 11: 169-174 ISSN 2283-5733 | DOI: 10.33393/grhta.2024.3107 ORIGINAL RESEARCH ARTICLE |
Valutazione dell’innovatività e negoziazione di prezzi e rimborso dei farmaci: raccomandazioni da un panel di esperti
Claudio Jommi![]() 1, Francesca Patarnello
1, Francesca Patarnello![]() 2, Cosetta Bianchi
2, Cosetta Bianchi![]() 3, Giuliano Buzzetti3
3, Giuliano Buzzetti3
Assessment of innovativeness, and price and reimbursement negotiation of medicines: recommendations of an expert panel
This paper illustrates the recommendations of a Working Group (WG) on the assessment of drugs innovativeness and the negotiation of price and reimbursement. The WG included researchers, institutions, clinicians, patient representatives and pharmaceutical companies.
The first part of the contribution summarizes the literature on drug pricing models, which was considered in the WG, and, in particular, the pricing criteria, the evaluation and negotiation processes, the management of the uncertainty of the evidence, the use of cross-reference pricing and price negotiation for new indications of existing drugs.
The second part illustrates the results of the WG with a focus on innovativeness assessment, value framework and price negotiation. The main recommendations of the WG are: to define more specific criteria for the identification of comparators and endpoints for macro therapeutic areas/settings; to produce guidelines on the use of indirect comparisons and studies supporting this evidence; to consider the drug value as the main driver of price and reimbursement negotiation; to maintain flexibility in the negotiation process, but, at the same time, to give greater structure and predictability in the assessment of value for money, with a more qualified role of cost-effectiveness and a range of threshold values for the incremental cost-effectiveness ratio; to selectively reintroduce Managed Entry Agreements and the Indication-based pricing model; to implement an early dialogue between the Italian Medicine Agency and the pharmaceutical companies in order to optimize the negotiation process, and a structured involvement of scientific societies and patient representatives.
Keywords: Assessment, Innovativeness, Medicines, Negotiation, Price and Reimbursement, Recommendations
Received: April 27, 2024
Accepted: June 14, 2024
Published online: July 15, 2024
Global & Regional Health Technology Assessment - ISSN 2283-5733 - www.aboutscience.eu/grhta
© 2024 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0).
Commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Introduzione
Il presente paper illustra i risultati di un Gruppo di Lavoro (GdL) che ha discusso il tema del sistema di valutazione dei nuovi farmaci e delle nuove indicazioni ai fini della valutazione delle richieste di innovatività e di Prezzo e Rimborso (P&R) in Italia. Il GdL è stato costituto nell’ambito della Sesta Edizione dei Seminari di Mogliano Veneto (“Diamo valore al percorso compiuto: temi irrisolti ed emergenti in un nuovo contesto regolatorio”), tenutasi il 21 e il 22 settembre 2023. Il GdL ha visto la partecipazione, come nelle precedenti edizioni dei Seminari di Mogliano, di soggetti istituzionali (Agenzia Italiana del Farmaco-AIFA, e Regioni) e stakeholder (clinici, referenti per i pazienti e imprese) (cfr. Ringraziamenti).
Il paper è diviso in due parti. Nella prima viene descritta in sintesi la letteratura sui modelli di pricing dei farmaci. Nella seconda vengono illustrati i risultati del GdL con un focus sul framework di valore e sul processo valutativo e negoziale dei prezzi.
Modelli e processi valutativi per il P&R dei farmaci
I modelli e i processi valutativi e negoziali per determinare il P&R dei medicinali sono stati ampiamente studiati in letteratura (1-3).
I prezzi sono definiti/regolati/negoziati sulla base di diversi approcci/modelli e in relazione a differenti prospettive. Nella prospettiva di chi valuta e acquista i farmaci, i principali driver sono: il valore terapeutico aggiunto rispetto ad alternative terapeutiche disponibili sul mercato, la coerenza tra valore comparativo e costo comparativo per paziente trattato (Value-Based Pricing, VBP) e la coerenza tra impatto sulla spesa e risorse disponibili (sostenibilità). Le imprese richiedono prezzi che consentano, insieme ai volumi, di generare ricavi tali da remunerare il capitale investito, vale a dire un margine sui costi sostenuti (Ricerca e Sviluppo, R&S, produzione, commercializzazione e altri costi amministrativi) (4).
Il VBP nei principali Paesi europei viene gestito sulla base di due modelli (Fig. 1) (4):
- il primo, presente nel Regno Unito, prevede che (i) il beneficio sia stimato in termini di aspettativa di vita ponderata per la qualità della stessa (Quality Adjusted Life Years, QALY) e che (ii) la coerenza tra costo incrementale e beneficio incrementale sia basata sul calcolo del rapporto incrementale di costo-efficacia (Incremental Cost-Effectiveness Ratio, ICER) e sulla verifica che tale rapporto sia inferiore a un valore soglia o range di valori soglia. Tale range è stato fissato in 20/30.000 sterline per anno di vita in perfetta salute, con clausole di salvaguardia per farmaci lanciati per patologie ultra-rare (con prezzi molto elevati) o con prognosi infausta (aspettativa di vita inferiore ai tre mesi), per le quali ottenere un aumento dei QALY importante è molto complesso;
- nel secondo, adottato dagli altri principali Paesi europei (oltre a Italia, Francia, Germania e Spagna), il valore aggiunto viene riconosciuto solo in caso di incremento dell’effetto terapeutico. La coerenza tra prezzo effettivo a carico del sistema pubblico e valore (terapeutico) aggiunto viene valutata in modo più o meno discrezionale: valore (terapeutico) aggiunto non esplicitamente misurato o valore aggiunto misurato attraverso sistemi di ranking, basati su entità relativa degli effetti, loro persistenza e qualità delle prove. Diverso è il ruolo assegnato ad altri elementi, quali le evidenze di costo-efficacia a integrazione della valutazione di coerenza tra prezzo e valore, la gravità della patologia target e il livello di bisogno (terapeutico) insoddisfatto.
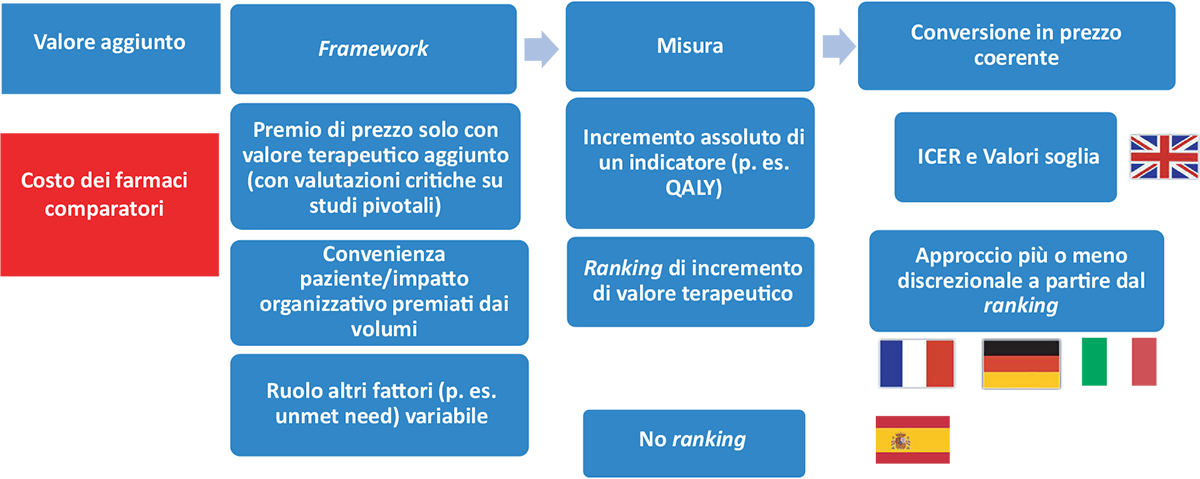
FIGURA 1 - VBP nei maggiori Paesi europei.
Fonte: Adattato da Jommi et al. (4).
QALY = Quality Adjusted Life Years; ICER = Incremental Cost-Effectiveness Ratio.
Il valore aggiunto non terapeutico, come la maggiore accettabilità della terapia per i pazienti o l’impatto sulle organizzazioni sanitarie, non rappresenta un elemento considerato nella negoziazione del prezzo (5,6), a meno che non vi siano evidenze che la maggiore accettabilità della terapia produca una maggiore compliance al trattamento (aderenza e persistenza) e che questa determini una maggiore efficacia e, quindi, un valore terapeutico aggiunto.
Come specificato sopra, la remuneratività del capitale investito rappresenta il driver delle richieste di prezzo da parte delle imprese. In realtà la fissazione dei prezzi sulla base dei costi sostenuti dalle imprese (metodo cost-plus) era piuttosto diffusa in Europa nella prima metà degli anni ’90. Tuttavia, tale metodo è stato gradualmente sostituito da un approccio VBP a causa della natura congiunta dei costi di R&S, vale a dire del fatto che tali costi sono sostenuti a vantaggio di tutti i Paesi e difficilmente allocabili sui singoli stati, perché premia i costi sostenuti e non l’esito della R&S e per la scarsa trasparenza e verificabilità dei dati di costo (7). In letteratura esistono diversi dati sui costi di R&S, mentre sono poche le prove sui costi di produzione e commercializzazione (8). Peraltro, con riferimento ai costi di R&S, due recenti revisioni di letteratura (9,10) hanno evidenziato un’importante variabilità nella stima di tali costi. Nella revisione di Schlander et al. (9), si riportano stime dei costi di R&S per molecola lanciata sul mercato comprese tra 161 milioni e 4,54 miliardi di dollari, con valori mediamente più alti per i farmaci oncologici (tra 944 e 4,54 miliardi di dollari) e decisamente più bassi per i farmaci a designazione orfana: 501 milioni di dollari per un orfano, rispetto a una media di 2,14 miliardi di dollari per un non orfano, secondo lo studio di Berdud et al. (11). Quest’ultimo risultato è da ascrivere al più basso numero di pazienti coinvolti negli studi e al frequente lancio di tali farmaci post Fase II. Le stime di costo includono i costi effettivi per lo sviluppo della molecola lanciata sul mercato e quelli generati dai fallimenti (per ogni molecola di successo), nonché la capitalizzazione di tali costi. Sono state mosse diverse critiche a tali stime, a partire dalla estrema variabilità degli esiti, dalla tendenziale bassa trasparenza degli studi e dalla scarsa considerazione di quanto tale ricerca clinica sia frutto non solo di investimenti di impresa ma anche di contributi pubblici (12-14). Tuttavia, per quanto le stime presentino un’elevata variabilità, diversi studi convergono su un tendenziale incremento dei costi di R&S verso valori vicini ai due miliardi di dollari per i farmaci per i quali viene realizzato uno sviluppo completo, vale a dire fino alla Fase III.
Come specificato sopra, nella prospettiva del pagatore, oltre alla coerenza tra valore e costo, è importante la sostenibilità rispetto alle risorse disponibili. In generale la sostenibilità non è un concetto assoluto, ma relativo, e dipende dai vincoli macroeconomici (essenzialmente debito pubblico rispetto al Prodotto Interno Lordo), dalla volontà politica di investire risorse pubbliche in sanità e dall’approccio di allocazione/gestione delle risorse più o meno orientato da tetti di spesa per fattori produttivi (silos) (7). Un incremento di spesa generato da un nuovo farmaco è tanto meno sostenibile, a parità di effetti sulla spesa pubblica, in presenza di vincoli macroeconomici stringenti, di una difficoltà di riallocazione della spesa pubblica da altri comparti al sistema sanitario e di tetti sulla spesa farmaceutica, aspetti che caratterizzano proprio il nostro Paese.
Oltre al tema dei modelli di pricing, la negoziazione dei prezzi dei farmaci presenta quattro ulteriori aspetti.
Il primo è rappresentato dall’uso dei prezzi di altri Paesi come riferimento per la determinazione di quelli nazionali (cross-reference pricing). Se tale pratica può integrare e, potenzialmente, facilitare il processo di negoziazione (15,16), non è apprezzata dalle imprese, nel caso i prezzi di riferimento esterni siano sistematicamente quelli più bassi, ed è complessa da applicare, vista la frequente presenza di sconti confidenziali. In alcuni contributi è stata sollevata la richiesta di una maggiore trasparenza dei prezzi al netto degli sconti (17,18), ma è stato anche evidenziato come questo potrebbe generare un ulteriore ritardo nel lancio (o il mancato lancio) in Paesi dove tali prezzi sono molto bassi (o gli sconti molto elevati), proprio per problemi di sostenibilità (19).
La presenza di incertezza sull’esito e sull’impatto finanziario di un farmaco al momento della negoziazione dei prezzi ha spinto molti Paesi a introdurre contratti di accesso al mercato o Managed Entry Agreement (MEA) (20). La letteratura sui MEA è ormai molto vasta e riporta la tassonomia di tali accordi (20-23) e valutazioni critiche sui MEA, in particolare quelli outcome-based, sistematizzate in un contributo di Antonanzas et al. (24) e che riguardano (i) il presunto burden amministrativo, (ii) la percezione che tali accordi vengano stipulati più per obiettivi di sostenibilità che per effettiva volontà di raccogliere dati real-life finalizzati a ridurre l’incertezza sul profilo di efficacia al lancio del nuovo prodotto/della nuova indicazione e (iii) la difficoltà, nel caso di accordi che prevedano studi post-marketing e una rinegoziazione successiva delle condizioni di accesso, di ridurre i prezzi e/o di escludere dalla rimborsabilità i farmaci con evidenze post-marketing non soddisfacenti (problemi di enforcement). Per quanto vi sia un trend di riduzione degli accordi outcome-based e di fatto gli stessi siano stati abbandonati in Italia, due recenti paper, partendo da tali valutazioni critiche, hanno suggerito dei modelli che possono, almeno parzialmente, superare le problematicità di tali accordi, salvaguardando il loro obiettivo, che è quello di condivisione del rischio di impatto non favorevole dei farmaci e di raccolta di dati real-world (25,26).
Va poi sottolineato come sia sempre più frequente la rinegoziazione del prezzo dei farmaci, per effetto dell’estensione delle indicazioni. La letteratura ha analizzato i modelli presenti nella pratica regolatoria/negoziale (27), rappresentati da modello brand e, conseguentemente, prezzo differenziato per indicazione, prezzo unico e sconti e/o MEA differenziati per indicazione (Indication-Based Pricing, IBP) e prezzo unico come media “ponderata” dei prezzi per indicazione (modello Blended Price). Diverse analisi hanno evidenziato i vantaggi dell’IBP, tra cui quello di rendere esplicito il prezzo effettivo per indicazione, e i limiti, tra cui la necessità di tracciare l’utilizzo dei farmaci per indicazione e un carico amministrativo la cui rilevanza dipende dalla complessità degli accordi. I limiti dell’IBP hanno portato diversi Paesi a utilizzare da sempre il modello Blended Price o, come l’Italia, a passare da un approccio IBP a uno di Blended Price. Un recente contributo ha analizzato, per l’Italia, la complessità di tali rinegoziazioni, evidenziando tempi maggiori rispetto alla prima indicazione e l’incremento dello sconto per effetto della rinegoziazione (28).
Infine, si sottolinea come il processo di negoziazione di P&R sia rilevante tanto quanto i criteri utilizzati, con riferimento in particolare a trasparenza, riproducibilità e allineamento tra requisiti di prova da parte dei pagatori e raccolta e analisi dei dati da parte dell’industria farmaceutica. Un quadro regolatorio che premia il valore terapeutico aggiunto attraverso un premio di prezzo e che rafforza la concorrenza sui prezzi per prodotti intercambiabili richiede un processo di P&R trasparente e riproducibile. A loro volta, trasparenza e riproducibilità dei processi negoziali chiariscono perimetro e criteri in base ai quali le imprese gestiscono la concorrenza. L’interazione precoce tra soggetti regolatori, pagatori e industria nel disegno degli studi clinici e tra pagatori e industria quando il prodotto viene approvato può migliorare il processo di valutazione e negoziazione, per un allineamento sulla valutazione (per esempio, su comparatori da utilizzare e legittimità del confronto indiretto) o per colmare le lacune informative (per esempio, sulla dimensione della popolazione target o sulla persistenza a lungo termine degli effetti dei medicinali) (29,30).
I risultati del GdL su innovatività e pricing
Come sopra specificato, il GdL ha affrontato il tema dei criteri e del processo di valutazione dell’innovatività dei farmaci/indicazioni e di negoziazione del P&R. La prima è valutata da AIFA (dalla CTS, Commissione Tecnico-Scientifica, prima della riforma, e dalla CSE, Commissione Scientifico-Economica, attualmente) sulla base di bisogno terapeutico, valore terapeutico aggiunto e qualità delle prove. Il valore terapeutico aggiunto è un elemento chiave anche per l’ottenimento di un premio di prezzo rispetto ai comparatori, non essendo possibile, ai sensi del DM 2/8/2019, prevedere un premio di prezzo in caso di assenza di valore terapeutico aggiunto. Non esiste però una regolamentazione specifica che gradui il premio di prezzo in funzione del ranking del valore terapeutico aggiunto, che invece è molto rilevante per ottenere l’innovatività.
Il GdL ha però riconosciuto che i due temi sono collegati. In primo luogo, come specificato sopra, l’innovatività è valutata sulla base del bisogno terapeutico, del valore terapeutico aggiunto e della qualità delle prove e almeno i primi due domini e, soprattutto il secondo, informano sul processo di negoziazione di P&R. L’acquisizione dell’innovatività piena, per quanto non formalmente prevista, rappresenta un elemento importante per ottenere un prezzo superiore alle alternative terapeutiche. Infine, la valutazione di innovatività e il sistema di pricing condividono alcune criticità (p. es., scelta delle alternative terapeutiche).
Dopo un ampio dibattito, è però emersa l’utilità di separare i due temi, riconducendo l’innovatività al suo ruolo, vale a dire di valutazione “regolatoria” finalizzata a individuare farmaci/indicazioni (i) da prioritizzare attraverso politiche specifiche, fondo dedicato e accesso immediato ai mercati regionali, quest’ultimo previsto anche in caso di innovazione potenziale, e (ii) da premiare, con la possibilità per le imprese di non applicare il 5%+5% di riduzione di prezzo e l’esclusione dei farmaci innovativi dal calcolo per la valutazione dello sfondamento dei tetti di spesa e, in particolare, dei tetti per acquisti diretti da parte delle aziende sanitarie.
Prima di discutere, sotto il profilo tecnico, della valutazione dell’innovatività, il GdL ha messo in evidenza come: (i) non vi siano evidenze sull’accesso immediato dei farmaci innovativi e che sarebbe opportuno verificare se ciò effettivamente avviene; (ii) la negoziazione si concluda spesso con la rinuncia al vantaggio di non applicare il 5%+5% di sconto; (iii) raramente l’innovatività condizionata si traduca in innovatività piena (è stato rintracciato solo un caso in cui questo si è verificato). Il GdL ha quindi proposto l’attivazione di un percorso più strutturato per la traduzione dell’innovatività condizionata in innovatività piena e di definizione del ruolo dei Real-World Data (RWD) in tale valutazione.
Un primo ambito di discussione del GdL è stato l’opportunità o meno di allargare la “candidabilità” all’innovatività in termini di:
- patologie eleggibili: attualmente è possibile richiedere l’innovatività se un farmaco è indicato per patologie con esito mortale, ospedalizzazioni ripetute, impatto rilevante su disabilità correlata alla qualità della vita;
- bisogno terapeutico e valore aggiunto, che oggi sono valutati sulla base della validità terapeutica delle alternative disponibili e del valore terapeutico aggiunto del farmaco/indicazione oggetto di valutazione rispetto alle alternative; non hanno invece rilievo né sulla dimensione del bisogno né su quella del valore aggiunto altri domini, come l’accettabilità delle terapie per i pazienti.
Il GdL non ha raggiunto un consenso sull’opportunità o meno di procedere a questa estensione, considerando anche che l’allargamento potrebbe generare un sensibile aumento dei farmaci candidabili (ed effettivamente riconosciuti come innovativi) con un rischio rilevante sulla “tenuta” del fondo per farmaci innovativi.
Con riferimento invece ai criteri e al processo di valutazione dell’innovatività, il GdL ha suggerito di prevedere criteri specifici di valutazione per macro-aree terapeutiche/setting, cercando di distinguere tra endpoint “hard” e altri indicatori di esito, analogamente a quanto già previsto per i farmaci oncologici. Il GdL ha peraltro evidenziato come sia opportuno declinare, anche per l’oncologia, gli endpoint più appropriati per setting (per esempio, la sopravvivenza non è l’endpoint più appropriato per un setting adiuvante). È stata poi espressa l’esigenza di aumentare la trasparenza sui criteri e il razionale della scelta delle alternative terapeutiche considerate, prioritizzando la migliore alternativa disponibile e l’alternativa maggiormente utilizzata nella pratica clinica, di fornire raccomandazioni sull’uso di confronti indiretti e studi a supporto, per evitare il più possibile un uso erratico degli stessi e approcci di tipo naïve, e di fornire un indirizzo su come dovrebbe avvenire la valutazione in assenza di valide alternative terapeutiche e su outcome “hard” non raggiunti negli studi clinici (per esempio, mancato raggiungimento degli obiettivi di sopravvivenza, scelto come endpoint secondario). Con riferimento al processo, il GdL ha suggerito di prevedere un coinvolgimento strutturato delle società scientifiche nella valutazione dell’entità del bisogno terapeutico, la definizione degli endpoint e la scelta delle alternative terapeutiche e delle associazioni di pazienti, quando rilevante, per esempio sulle evidenze di qualità della vita correlata allo stato di salute.
Rispetto alla negoziazione di P&R, date l’ampiezza e complessità dell’argomento, il GdL si è concentrato su quattro elementi: l’individuazione delle alternative terapeutiche, il legame tra valore e prezzo, i contratti di accesso e la rinegoziazione a seguito dell’estensione delle indicazioni.
Con riferimento alle alternative terapeutiche, si è condivisa la stessa raccomandazione fatta per l’innovatività, vale a dire di aumentare la trasparenza su criteri di scelta delle alternative, limitandole a quelle autorizzate per l’indicazione e il rimborso e selezionando o prioritizzando nel confronto la migliore alternativa disponibile e/o quella di maggiore uso (che non necessariamente è un farmaco) o quella per la quale la qualità delle prove è migliore. In previsione dell’applicazione del Regolamento UE sull’HTA (Regolamento EU 2021/2282), il GdL ha poi raccomandato un ruolo fortemente proattivo di AIFA nel supporto all’individuazione delle alternative terapeutiche nell’ambito del Joint Clinical Assessment.
Rispetto al pricing, il GdL ha in primo luogo richiamato l’opportunità che la negoziazione di P&R abbia come primi driver il valore e la coerenza tra valore e costo in termini comparativi e poi l’impatto sulla spesa.
Il GdL ha poi riconosciuto che la flessibilità del sistema negoziale abbia garantito la rimborsabilità dei farmaci in Italia in misura maggiore rispetto a diversi Paesi europei (31), ma che sia opportuno pensare a una struttura e a una prevedibilità maggiori nel collegamento tra valore e costo della terapia. Un primo passo potrebbe essere la pubblicazione del ranking del valore terapeutico aggiunto e del bisogno terapeutico per tutti i nuovi farmaci e le indicazioni e non solo per quelli per i quali è stato richiesto il riconoscimento dell’innovatività. Un secondo passo suggerito dal GdL è quello di dare un maggior peso, pur mantenendo un approccio multi-criterio, alla costo-efficacia, con range di valori soglia per il rapporto incrementale di costo-efficacia. Il GdL ha poi suggerito di esplicitare quando l’analisi di costo-efficacia può avere un maggiore ruolo informativo/di supporto, vale a dire nei casi di valore terapeutico aggiunto significativo e in assenza di alternative terapeutiche.
Rispetto a domini del valore da considerare, il GdL ha proposto di valutare la possibilità che un valore aggiunto riferito a dimensioni “non direttamente terapeutiche” (come la migliore accettabilità di una terapia per il paziente) venga considerato nella negoziazione, almeno nel contenimento degli eventuali sconti sul prezzo richiesto, non essendo possibile, ai sensi del DM 2/8/2019, prevedere un premio di prezzo in caso di assenza di valore terapeutico aggiunto.
Il GdL ha poi raccomandato un’interazione precoce, anche attraverso scoping meeting tra AIFA e imprese, sulle evidenze a supporto del valore, considerando tutti gli aspetti ritenuti critici, incluse la scelta delle alternative terapeutiche e la stima del costo-terapia delle alternative al farmaco oggetto di negoziazione.
Infine, sui contratti di accesso e rinegoziazioni, il GdL ha raccomandato di riattivare i MEA outcome-based basati su registri di farmaci, evitando però l’errore commesso nel passato, vale a dire di utilizzare sistematicamente i MEA come strumento per superare un’impasse negoziale o come strumento di pura gestione della “sostenibilità”, limitandoli ai casi specifici in cui vi sia un’elevata incertezza su dimensione e durabilità dell’efficacia dei farmaci. Il GdL ha invece espresso dei dubbi sulla praticabilità di approcci alternativi, basati su programmi strutturati di raccolta dati ad hoc e sulla rinegoziazione dei prezzi in relazione alle evidenze raccolte (Coverage with Evidence Development). Tali studi sarebbero molto complessi da gestire, per quanto abbiano più senso quando i trattamenti oggetto di valutazione sono terapie di lunga durata per patologie croniche e quando la valutazione dell’esito si basa su endpoint multipli.
In parallelo è stato suggerito di ripristinare un approccio di IBP, almeno nel caso in cui la nuova indicazione approvata abbia un alto valore per il Servizio Sanitario Nazionale (per esempio, se viene valutata come innovativa). L’approccio IBP, oltre a garantire una maggiore aderenza del prezzo al valore del farmaco nell’indicazione approvata e rimborsata, consente di identificare il costo effettivo del farmaco per l’indicazione, costo che può rappresentare un termine di confronto per nuovi farmaci nella stessa indicazione. Se l’IBP non fosse possibile, sarebbe almeno opportuno esplicitare i criteri di ponderazione nell’applicazione del blended price, vale a dire quanto contano il valore e la dimensione della popolazione target nella ponderazione del prezzo medio.
Conclusioni
Il presente paper ha descritto i risultati della discussione di un GdL multi-disciplinare e multi-stakeholder finalizzata a proporre raccomandazioni sul sistema di valutazione delle richieste di innovatività e di negoziazione del P&R in Italia.
Pur riconoscendo l’importanza di avere definito un percorso ad hoc per la valutazione dell’innovatività e l’utilità di avere un sistema flessibile di negoziazione del P&R, il GdL ha evidenziato alcuni limiti dell’attuale assetto. Il fil rouge delle raccomandazioni è un aumento (i) della strutturazione e della trasparenza del processo, con riferimento in particolare alla definizione dei comparatori per la valutazione del valore aggiunto dei farmaci, alla modalità di applicazione del VBP e al ruolo della costo-efficacia nella negoziazione del P&R e (ii) dell’interazione tra AIFA e stakeholder, in particolare società scientifiche e associazioni di pazienti.
Tali raccomandazioni sono in linea con quelle di un pool di 39 esperti di accesso al mercato, provenienti da istituzioni pubbliche, ricerca e consulenza, raccolte attraverso un questionario strutturato (Jommi et al., 2023), riferite più in generale al sistema di pricing nei Paesi in cui il prezzo è regolamentato. Gran parte dei rispondenti ha evidenziato l’opportunità di (i) regolare il prezzo dei farmaci sulla base di un VBP, basandosi su un approccio multi-criterio, con l’eventuale supporto della costo-efficacia, e di MEA per la gestione dell’incertezza e (ii) aumentare la trasparenza dei processi negoziali (più che dei prezzi netti), valorizzando l’interazione con stakeholder e prevedendo contatti preliminari tra soggetti valutatori sugli aspetti più critici (entità del bisogno insoddisfatto, comparatori e dimensione della popolazione target).
L’auspicio è che tali raccomandazioni possano essere prese in considerazione, visto il momento dinamico caratterizzato dall’implementazione della riforma di AIFA e dall’applicazione, prevista da gennaio 2025, del Regolamento UE sull’HTA.
Acknowledgements
Un ringraziamento a tutto il team di Dephaforum. Si ringraziano tutti i partecipanti al Gruppo di Lavoro, che hanno condiviso le raccomandazioni presentate: Angelo Avogaro - Università degli Studi di Padova, Federica Basso - Advanz Pharma, Giovanni Corrao - Università degli Studi di Milano-Bicocca, Giorgio Corsico - Amgen, Francesco De Lorenzo - Presidente F.A.V.O., Mauro Di Gesù - Sanofi, Guido Di Donato - Pfizer, Cristina Dondoni - Gilead, Filippo Drago - Università degli Studi di Catania, Sebastian Iovan - Eli Lilly, Roberta Lo Muto - Biogen, Giuseppe Mancia - Università degli Studi di Milano-Bicocca, Mario Mangrella - Italfarmaco, Patrizia Olivari - Ipsen, Roberto Pala - Menarini, Stefano Palcic - Azienda Sanitaria Universitaria Giuliano Isontina, Marcello Pani - Policlinico Universitario A. Gemelli IRCCS, Luca Pernarella - AbbVie, Pasquale Perrone Filardi - Università degli Studi di Napoli Federico II, Lara Pippo - CSL Behring, Andrea Pitrelli - Shionogi, Stefania Pulimeno - Teva, Massimo Riccaboni - IMT School for Advanced Studies Lucca, Viviana Ruggieri - Servier, Pierluigi Russo - AIFA, Marco Zibellini - Farmindustria.
Disclosures
Conflict of interest: CJ reported serving as an advisory board member and a paid speaker for Abbvie, Amgen, AstraZeneca, Bristol Myers Squibb, CSL Behring, Gilead, Incyte, Merck Sharp & Dohme, Roche, Sanofi, Takeda, outside the submitted work. FP is an employee of AstraZeneca.
Financial support: This study received no grant from any funding agency in the public, commercial, or not-for-profit sectors.
Bibliografia
- 1. Garattini L, Cornago D, De Compadri P. Pricing and reimbursement of in-patent drugs in seven European countries: a comparative analysis. Health Policy. 2007;82(3):330-339. CrossRef PubMed
- 2. Kleijnen S, Lipska I, Leonardo Alves T, et al. Relative effectiveness assessments of oncology medicines for pricing and reimbursement decisions in European countries. Ann Oncol. 2016;27(9):1768-1775. CrossRef PubMed
- 3. Panteli D, Arickx F, Cleemput I, et al. Pharmaceutical regulation in 15 European countries review. Health Syst Transit. 2016;18(5):1-122. PubMed
- 4. Jommi C, Armeni P, Costa F, Bertolani A, Otto M. Implementation of Value-based Pricing for Medicines. Clin Ther. 2020;42(1):15-24. CrossRef PubMed
- 5. Vogler S, Paris V, Ferrario A, et al. How Can Pricing and Reimbursement Policies Improve Affordable Access to Medicines? Lessons Learned from European Countries. Appl Health Econ Health Policy. 2017;15(3):307-321. CrossRef PubMed
- 6. Garner S, Rintoul A, Hill SR. Value-Based Pricing: L’Enfant Terrible? PharmacoEconomics. 2018;36(1):5-6. CrossRef PubMed
- 7. Jommi C, Bertolani A, Armeni P, Costa F, Otto M. Pharmaceutical pricing and managed entry agreements: an exploratory study on future perspectives in Europe. Health Policy Technol. 2023;12(3):100771. CrossRef
- 8. Suresh P, Basu PK. Improving Pharmaceutical Product Development and Manufacturing: Impact on Cost of Drug Development and Cost of Goods Sold of Pharmaceuticals. J Pharm Innov. 2008;3(3):175-187. CrossRef
- 9. Schlander M, Hernandez-Villafuerte K, Cheng CY, Mestre-Ferrandiz J, Baumann M. How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. PharmacoEconomics. 2021;39(11):1243-1269. CrossRef PubMed
- 10. Rennane S, Baker L, Mulcahy A. Estimating the Cost of Industry Investment in Drug Research and Development: A Review of Methods and Results. Inquiry. 2021;58. CrossRef PubMed
- 11. Berdud M, Drummond M, Towse A. Establishing a reasonable price for an orphan drug. Cost Eff Resour Alloc. 2020;18(1):31. CrossRef PubMed
- 12. Light DW, Warburton R. Demythologizing the high costs of pharmaceutical research. Biosocieties. 2011;6(1):34-50. CrossRef
- 13. Schippers I, de Haan E, Cowan R. Overpriced - Drugs Developed with Dutch Public Funding. SOMO- Report 2019 Amsterdam. Online, (Accessed April 2024)
- 14. Schmidt L; Wild C. Assessing the Public and Philanthropic Financial Contribution to the Development of New Drugs: A Bibliographic Analysis. Sci. Technol. Public Policy 2020, 4(1), 8-14. CrossRef
- 15. Rand LZ, Kesselheim AS. International reference pricing for prescription drugs: a landscape analysis. J Manag Care Spec Pharm. 2021;27(9):1309-1313. CrossRef PubMed
- 16. Gill J, Fontrier AM, Kyriopoulos D, Kanavos P. Variations in external reference pricing implementation: does it matter for public policy? Eur J Health Econ. 2019;20(9):1375-1397. CrossRef PubMed
- 17. Morgan SG, Bathula HS, Moon S. Pricing of pharmaceuticals is becoming a major challenge for health systems. BMJ. 2020;368:l4627. CrossRef PubMed
- 18. Vogler S, Zimmermann N, Habl C, Piessnegger J, Bucsics A. Discounts and rebates granted to public payers for medicines in European countries. South Med Rev. 2012;5(1):38-46. PubMed
- 19. Riccaboni M, Swoboda T, Van Dyck W. Pharmaceutical net price transparency across european markets: insights from a multi-agent simulation model. Health Policy. 2022;126(6):534-540. CrossRef PubMed
- 20. Wenzl M, Chapman S. Performance-based managed entry agreements for new medicines in OECD countries and EU member states: How they work and possible improvements going forward. OECD Health Working Papers, 2019, No. 115, OECD Publishing, Paris. Online (Accessed April 2024)
- 21. Morel T, Arickx F, Befrits G, et al. Reconciling uncertainty of costs and outcomes with the need for access to orphan medicinal products: a comparative study of managed entry agreements across seven European countries. Orphanet J Rare Dis 2013;24;8:198. CrossRef PubMed
- 22. Carlson JJ, Chen S, Garrison LP Jr. Performance-Based Risk-Sharing Arrangements: An Updated International Review. PharmacoEconomics. 2017;35(10):1063-1072. CrossRef PubMed
- 23. Jommi C. Chapter 4 - Managed Entry Agreements and High Cost Medicines (European Perspective), in Zaheer- Ud-Din Babar (ed.). Equitable Access to High-Cost Pharmaceuticals, Springer, London.2018; 35-49. CrossRef
- 24. Antonanzas F, Juárez-Castelló C, Lorente R, Rodríguez-Ibeas R. The Use of Risk-Sharing Contracts in Healthcare: Theoretical and Empirical Assessments. PharmacoEconomics. 2019;37(12):1469-1483. CrossRef PubMed
- 25. Whittal A, Jommi C, De Pouvourville G, et al. Facilitating More Efficient Negotiations for Innovative Therapies: A Value-Based Negotiation Framework – Corrigendum. Int J Technol Assess Health Care. 2022;38(1):e43. CrossRef PubMed
- 26. Xoxi E, Rumi F, Kanavos P, et al. A Proposal for Value-Based Managed Entry Agreements in an Environment of Technological Change and Economic Challenge for Publicly Funded Healthcare Systems. Front Med Technol. 2022;4:888404. CrossRef PubMed
- 27. Campillo-Artero C, Puig-Junoy J, Segú-Tolsa JL, Trapero-Bertran M. Price Models for Multi-indication Drugs: A Systematic Review. Appl Health Econ Health Policy. 2020;18(1):47-56. CrossRef PubMed
- 28. Rossini EE, Galeone C, Lucchetti C, Jommi C. From Indication-Based Pricing to Blended Approach: Evidence on the Price and Reimbursement Negotiation in Italy. PharmacoEconom Open. 2024;8(2):251-261. CrossRef PubMed
- 29. Wang T, McAuslane N, Gardarsdottir H, Goettsch WG, Leufkens HGM. Building HTA insights into the drug development plan: current approaches to seeking early scientific advice from HTA agencies. Drug Discov Today. 2022;27(1):347-353. CrossRef PubMed
- 30. Wang T, McAuslane N, Liberti L, Leufkens H, Hövels A. Building Synergy between Regulatory and HTA Agencies beyond Processes and Procedures-Can We Effectively Align the Evidentiary Requirements? A Survey of Stakeholder Perceptions. Value Health. 2018;21(6):707-714. CrossRef PubMed
- 31. EFPIA. (2023). Patients W.A.I.T. Indicator 2022 Survey. Online (Accessed April 2024)