|
Glob Reg Health Technol Assess 2024; 11: 51-54 ISSN 2283-5733 | DOI: 10.33393/grhta.2024.3025 POINT OF VIEW |
Regolamento HTA: come si sta muovendo l’Italia?
HTA regulation: how is Italy moving?
Within the European evolutionary framework concerning citizens’ health, the focus shifts to the new HTA regulation, set to alter processes and influence decision-making at the individual country level regarding reimbursement and pricing. The ambitious goal of achieving faster and more uniform access will significantly reshape the evaluative model stabilized over the years, characterized by the creation of different rules and processes among member states and diverse output timelines across the countries.
The imminent adoption of a more collaborative process necessitates member countries and companies to address several steps to ensure a smooth transition without penalizing the unique aspects of individual countries or impeding real access. Italy, actively participating in European preparatory activities with AIFA, faces challenges in adapting its formal process due to AIFA’s ongoing reform since November 2022, which is not yet materialized.
National-level companies heavily depend on leadership and involvement rules from their HTA agencies, with the risk that agencies with more established engagement models with external stakeholders may present themselves on January 12, 2025, the start date of the new process for oncological and ATMP drugs, better prepared and may excel in designing scoping meetings and in formulating PICO.
Therefore, the activation of a country culture of HTA and a robust and extended “readiness” phase involving all stakeholders are desirable, along with the establishment of collaboration networks with universities and scientific societies to make valuable knowledge and qualified resources more readily available, crucial element for the delicate transition from the old to the new evaluative system.
Keywords: Early Access, Health Technology Assessment, Pricing and Reimbursement, Unmet need
Received: January 25, 2024
Accepted: February 5, 2024
Published online: March 5, 2024
Global & Regional Health Technology Assessment - ISSN 2283-5733 - www.aboutscience.eu/grhta
© 2024 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0).
Commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Premessa
La riforma della legislazione Farmaceutica e l’HTA regulation condividono come elemento centrale il proposito di costruire un framework che porti i pazienti in tutta l’UE ad accedere tempestivamente e in modo equo ai medicinali. L’impegno dell’Unione Europea sulla politica sanitaria trae origine dalle disposizioni in materia di salute e sicurezza e si è poi sviluppato come conseguenza della libera circolazione delle persone e dei beni nel mercato interno. Oggi pensiamo che il COVID-19 sia stato l’episodio più rilevante per dare maggiore priorità alla sanità pubblica nell’UE, ma negli anni ’90 è stata la crisi dell’encefalopatia spongiforme bovina (“il morbo della mucca pazza”) a dare priorità alla salute e alla protezione dei consumatori nell’agenda politica dell’UE, con una consapevolezza che ha portato poi alla nascita di due importanti istituzioni europee, l’Agenzia europea per i medicinali (EMA) (1993) e il Centro europeo per la prevenzione e il controllo delle malattie (ECDC) (2004), fino ad arrivare al lancio del programma EU4health, nel 2021-2027 (1), una visione di un’Unione europea più sana; in questo programma sono delineati tre importanti pilastri: a) la risposta all’emergenza COVID-19 e il rafforzamento della resilienza dell’UE alle minacce sanitarie transfrontaliere; b) il piano europeo di lotta contro il cancro; c) la strategia farmaceutica per l’Europa.
Ribadire il concetto che all’interno dell’UE si debba lavorare per una maggiore equità e uniformità di accesso ai medicinali è un obiettivo tanto importante quanto ambizioso se pensiamo al fatto che l’UE è tutt’altro che omogenea in termini di ricchezza e di potere d’acquisto dei cittadini, di demografia, di policy e di caratteristiche dei sistemi sanitari e che il PIL pro-capite varia nei 28 paesi UE in modo significativo.
Così, il 31 gennaio 2018, la Commissione Europea fa un Comunicato Stampa “Valutazione delle tecnologie sanitarie nell’UE: la Commissione propone di rafforzare la cooperazione tra gli Stati membri” nel quale sostiene che la proposta è volta a promuovere la cooperazione tra gli Stati membri dell’UE in merito alla valutazione delle tecnologie sanitarie, a una maggiore trasparenza e a poteri più ampi ai pazienti, garantendo loro l’accesso a informazioni sul valore clinico aggiunto di nuove tecnologie che potrebbero potenzialmente recare loro beneficio, un vantaggio, secondo la CE, anche per le autorità nazionali, che potranno formulare politiche per i propri sistemi sanitari sulla base di evidenze più solide, e per i “fabbricanti”, che non dovranno più adeguarsi a procedure nazionali differenti.
Nello stesso comunicato, il Vicepresidente Jyrki Katainen dichiarava che: “Rafforzare la cooperazione in materia di valutazione delle tecnologie sanitarie a livello dell’UE stimola l’innovazione e migliora la competitività delle imprese di tale comparto. Il settore dell’assistenza sanitaria costituisce una parte fondamentale della nostra economia, rappresentando circa il 10% del PIL dell’Unione. Il quadro normativo che proponiamo apporterà benefici ai pazienti in tutta l’Europa, incoraggiando nel contempo l’innovazione, favorendo la diffusione di innovazioni di elevata qualità nel settore medtech e migliorando la sostenibilità dei sistemi sanitari dell’UE”.
La proposta di regolamento relativo alla valutazione delle tecnologie sanitarie, che riguarda i nuovi medicinali e alcuni nuovi dispositivi medici, intende stimolare la cooperazione tra Stati membri con la creazione e l’utilizzo di strumenti, metodologie e procedure comuni e definisce anche quattro settori principali di collaborazione: 1) valutazioni cliniche congiunte incentrate sulle tecnologie sanitarie più innovative, dall’impatto potenzialmente più significativo sui pazienti; 2) consultazioni scientifiche congiunte grazie alle quali gli sviluppatori possono chiedere la consulenza delle autorità di valutazione delle tecnologie sanitarie; 3) individuazione delle tecnologie sanitarie emergenti al fine di riconoscere precocemente le tecnologie promettenti; 4) proseguimento della cooperazione volontaria in altri settori. Come è noto, lascia la responsabilità della valutazione degli aspetti non clinici (per esempio, economici, sociali ed etici) e le decisioni in materia di fissazione dei prezzi e di rimborso ai singoli paesi dell’UE.
Arrivare a una “valutazione congiunta”, la famosa Joint Clinical Assessment (JCA) è quindi un vantaggio per il cittadino e per il “fabbricante”, e può alleggerire l’impatto valutativo dei singoli stati membri e delle loro agenzie di HTA. Di fatto, questo “vantaggio” porterebbe, per i paesi con una maggiore struttura nell’HTA a una maggiore complessità “politica”.
Una valutazione unica significa, banalmente, poter gestire le decisioni sulla rimborsabilità o meno SOLO sulla parte di unmet need o economico-organizzativa. Questo significa che, data una certa JCA, la scelta del paese non potrà essere, se non in casi eccezionali e documentati, una diversa valutazione comparativa, ma un’appraisal generale dell’agenzia di HTA del paese membro che, tenuto conto di quella valutazione congiunta, ne valuta l’idoneità per il rimborso.
Dobbiamo quindi pensare a una valutazione che sia: adeguata in termini di metodologia (HTA), disponibile nello stesso momento e inclusiva di tutti gli elementi distintivi rilevabili nei singoli paesi (p. es., Standard of care).
È con questa prospettiva che, a mio avviso, va studiata l’implementazione della riforma HTA in Italia, a ormai circa 13 mesi dalla sua effettività almeno per una parte, non secondaria, di farmaci (oncologici e ATMP), per comprendere che cosa potrebbe cambiare e come prepararci.
Il cambiamento si presenta come una vera rivoluzione culturale:
- il passaggio formale a un sistema in cui la valutazione scientifica si concentra sul concetto di relative effectiveness e su un PICO generato in modo collaborativo con gli altri stati membri;
- lo sviluppo e la conseguente adozione di Linee Guida, template e procedure uniche;
- la produzione di due risultati importanti e impegnativi per lo svolgimento delle attività degli stati membri per il P&R come il Joint Scientific Consultation e il Joint Clinical Assessment.
L’inclusione, reale, della prospettiva di pazienti e di esperti nello scoping e nel PICO, cosa che potrebbe mettere molto in difficoltà alcune agenzie di HTA e magari anche alcuni Health Technology Developers (HTD).
Cosa cambia per l’HTA body di uno stato membro?
Chiaramente la presenza di un processo “centralizzato” che produce una documentazione strutturata sulla relative effectiveness, la cosiddetta JCA, potrebbe prevenire uno dei punti più critici delle valutazioni scientifiche dei paesi vale a dire il fatto che ciascuno abbia creato e consolidato nel tempo processi valutativi diversi, dal punto di vista sia delle priorità/fast track che della comparazione che dell’inclusione di dimensioni non esclusivamente cliniche nelle valutazioni, con i quali ha trovato un proprio equilibrio tra “discrezionalità” delle valutazioni tecnico-scientifiche e condizioni di rimborso. Questo significa non poter lavorare sulla componente clinica comparativa per raggiungere degli obiettivi di prezzo.
Inoltre lo stato membro deve essere in grado di muoversi nel processo “joint” in modo adeguato e lo dovrà fare probabilmente con risorse di maggiore seniority in grado di influenzare l’HTA board. Queste a loro volta dovranno essere affiancate da gruppi di lavoro che producano valutazioni e analisi. Questo significa una programmazione parallela e tarata sui processi europei che potrebbe essere abbastanza dispendiosa.
E come gestire l’evoluzione normativa e dei regolamenti delle diverse agenzie HTA? Questi saranno ancora validi o dovranno essere nel frattempo emendati?
Infine, lo stato membro potrà condividere “volontariamente” altre informazioni con gli altri stati membri. Chi prevarrà? Come verranno gestite eventuali discrepanze? In altre parole, che politiche intraprendere e trasferire ai cittadini circa le possibili differenze nel processo di valutazione tra paesi? Questo significa essere più esposti alle critiche in relazione alle aspettative generate dalla legislazione farmaceutica e dall’HTA regulation stessa di maggiore equità e tempestività!
Certamente l’Horizon scanning farà la differenza. Probabilmente andrebbe esteso dall’analisi attuale delle molecole approvate o in approvazione a una valutazione -36, -24, -12 mesi, meglio se con raggruppamenti per tematiche critiche (per esempio combinazioni). L’HS deve infatti alimentare la fase di consultazione precoce che è fondamentale per avviare un dialogo tra tutti gli attori, compresa l’industria, al fine di poter ricevere advice su come poter migliorare gli studi, prima di arrivare alla JCA (quando sarebbe troppo tardi).
Il coinvolgimento di pazienti, di esperti esterni e di stakeholder nell’Horizon Scanning e nella gestione del COI
Sia a livello europeo che nei singoli stati membri, anche in accordo con quelli che sono i principi e gli obiettivi chiave di questa riforma (ricordiamo: maggiore rilevanza dei pazienti, maggiore accesso all’innovazione), il tema del coinvolgimento di esperti e stakeholder è un elemento chiave.
Identificare, in ogni paese, questo processo, in modo tale da potere, già al momento dell’Horizon Scanning, acquisire informazioni importanti per esempio sulle priorità dei pazienti o sugli elementi critici dei percorsi di cura da parte degli operatori sanitari, risulta un elemento chiave ai fini della preparazione e del contributo nello scoping, nel PICO e nella JCA. Lo schema proposto da EUnetHTA (Fig. 1) (2) rimanda alle regolamentazioni e alle prassi nazionali per quanto riguarda gli input degli esperti.
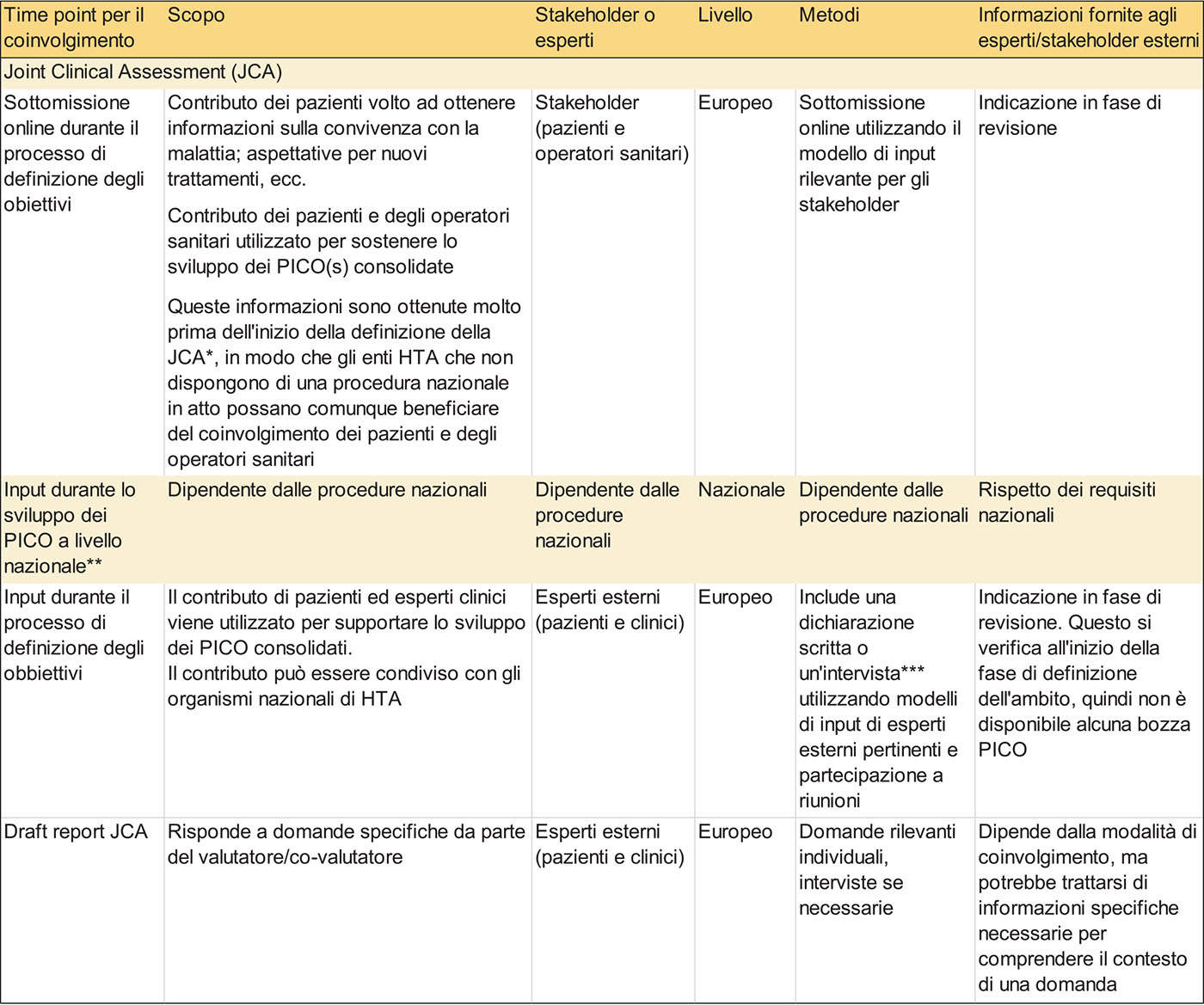
FIGURA 1 - Overview del coinvolgimento di esperti esterni nei risultati riportati da EUnetHTA 21. Tradotta da European Network for Health Technology Assessment (EUnetHTA). Guidance Document. D7.2 (2).
*Nota: i modelli di input degli esperti esterni verranno utilizzati da 1 a 3 mesi prima dell’avvio del processo di definizione degli obiettivi della JCA. Il processo per raccogliere questi input durerà circa 1 mese.
**Nota: le aree ombreggiate rappresentano il coinvolgimento nazionale che potrebbe essere svolto a livello nazionale, secondo le procedure nazionali. Se l’HTAb (Health Technology Assessment body) nazionale richiedesse il contributo di esperti e stakeholder nazionali, questo input dovrebbe essere riflesso nella posizione individuale dell’HTAb (Health Technology Assessment body) in un JSC e nel PICO nazionale per la JCA. A differenza del coinvolgimento degli esperti europei, le opinioni degli esperti o degli stakeholder nazionali non sono direttamente incluse nel rapporto/appendice della JCA o nella raccomandazione scritta finale/appendice del JSC.
***Nota: un’intervista avrà luogo tra esperto esterno e valutatore/co-valutatore/segreteria. Dopo l’intervista, verrà condiviso un riassunto con l’esperto esterno per la validazione dei contenuti.
Certamente l’Horizon Scanning con il contributo degli esperti esterni farà la differenza e sicuramente la partecipazione di interlocutori effettivamente esperti e/o rappresentativi, compatibilmente con le regole del COI, sarà un elemento chiave per le singole agenzie HTA.
Nel caso italiano, la realizzazione dell’attività di HS potrebbe coincidere con la messa a punto di una consultazione pubblica con esperti e portatori di interesse e rappresentare il primo grande momento di attuazione del processo di HTA, che considera tale coinvolgimento come elemento chiave della valutazione.
In termini di tempistiche probabilmente andrebbe esteso dall’analisi attuale delle molecole approvate o in approvazione a una valutazione -36, -24, -12 mesi, meglio se con raggruppamenti.
Esiste un tema di capacity determinato o aggravato dall’HTA?
Prima di portare i suoi importanti frutti in termini di omogeneità e di riduzione delle duplicazioni, l’adattamento alle nuove regole peserà in termini di capacity sulle agenzie HTA. I paesi, come l’Italia, che non hanno strutturato un’alleanza con un network di Università e centri di ricerca nell’HTA si troveranno in seria difficoltà nella competizione con paesi europei con strutture più capienti e con maggiori risorse. Questo network va costruito in modo formale con accordi trasparenti e con metodologie chiare e potrebbe veramente fare la differenza nella capacity e anche nella qualità delle valutazioni, consentendo in modo più agile anche un dialogo, ben normato, con tutti gli stakeholder del processo.
È sempre nella declaratoria iniziale di EU4Health che troviamo il riferimento al mondo universitario quando si dichiarano le “parti interessate”, che, scrive l’Europa, comprendono i rappresentanti della società civile e delle associazioni di pazienti, il mondo universitario e le organizzazioni degli operatori sanitari, i quali forniscono contributi sulle priorità e sugli orientamenti strategici e sulle esigenze da affrontare attraverso il programma di lavoro annuale.
La rete degli esperti di area clinica e farmaceutica e di metodologia (economia, statistica, modellistica) non può che essere trovata e costruita attraverso il rapporto con le università, i centri di conoscenza, gli scambiatori di esperienze con il mondo internazionale e i formatori dei prossimi esperti. Riflettere sulle migliori modalità per costruire questa relazione è un elemento chiave per il nostro futuro e per superare uno dei nodi più importanti, vale a dire quello dell’incremento rapido della capacity in AIFA con personale motivato a intraprendere una carriera pubblica in un ambito così delicato e dove la competizione tra pubblico e privato e tra Italia e estero è, per i giovani più brillanti e competenti, molto elevata.
Anche per la marketing company, la filiale di una multinazionale, ci saranno dei cambiamenti
Anche in questo caso, specialmente per i paesi come l’Italia, nei quali non c’è, tranne che in casi eccezionali, un dialogo precoce con AIFA per la messa a punto del dossier di P&R della nuova molecola/indicazione, sarà necessaria una maggiore preparazione da parte delle unità di V&A/HTA/HE delle affiliate e degli headquarter. Mi posso aspettare che in qualche modo si “raddoppi” l’intervento di questi team:
- nella preparazione e nella messa a disposizione degli HQ di tutto ciò che concorre a rendere la JCA la migliore e la più completa possibile a partire dalle analisi delle alternative terapeutiche e dalla posizione delle associazioni di pazienti;
- nella messa a punto delle strategie post-JCA per consentire quindi il completamento con la parte HTA di costi, impatti organizzativi, valorizzazione e prezzo.
Con la partenza prevista per il 1o gennaio 2025 dovremmo avere già tutti alla mano la lista possibile dei farmaci che saranno oggetto del nuovo percorso. Questi dossier conviveranno con tutti gli altri.
Difficile immaginare dove si possa “intoppare” la macchina, dato che siamo a un anno dalla riforma dell’AIFA e che ancora non abbiamo né la struttura di funzionamento della nuova AIFA né le qualifiche dei responsabili dei ruoli dirigenziali e consultivi e dato che non si è aperta una consultazione pubblica sulla trasformazione del decreto del 2 agosto 2019 che a oggi rappresenta il framework normativo di valutazione e che certamente poco si adatta sia come spirito che come passaggi metodologici alla riforma europea.
Una proposta
Una proposta è quella di costruire il più velocemente possibile un working group misto, con rappresentanti di AIFA, delle regioni, del Ministero, dell’Università, dei pazienti, delle società scientifiche e delle aziende.
In pratica, un’esperienza di “HTA readiness” che ci aiuti a costruire le migliori squadre per gli stakeholder coinvolti, che costruisca un percorso valutativo “mock” che possa “allenare” i diversi gruppi e che simuli le interazioni prima di gennaio 2025, identificando dall’inizio gli elementi critici.
Acknowledgements
We wish to thank Dr. Arturo Mangani for his editorial review of this article.
Disclosures
Conflict of interest: The Authors declare no conflict of interest.
Funding and financial support: FP is an employer of AstraZeneca Spa. The views expressed in this article are the author’s own views and do not necessarily reflect the company’s view.
Bibliografia
- 1. Commissione Europea. Programma EU4Health 2021-2027 – una visione per un’Unione europea più sana. 2024. Online (accessed January 2024)
- 2. European Network for Health Technology Assessment (EUnetHTA). Guidance Document. D7.2 – Guidance on patient & Healthcare professional involvement. Version 1.0, 04.04.2023. p21 Online (accessed January 2024)