|
Glob Reg Health Technol Assess 2023; 10: 79-88 ISSN 2283-5733 | DOI: 10.33393/grhta.2023.2610 ORIGINAL RESEARCH ARTICLE |
Time to market access in Italia: durata del processo di P&R per i farmaci per le malattie rare
Time to market access in Italy: duration of the P&R process for rare disease drugs
Objective: This paper aims to investigate the duration of the pricing & reimbursement (P&R) procedures submitted in Italy by pharmaceutical marketing authorization holders (MAH) for drugs indicated for rare diseases.
Methods: All the data used in this analysis were publicly available on different sources of the Italian Ministry of Health, the Italian Medicines Agency (AIFA) and other official websites. The information was systematically collected to investigate the timeline (days) needed to complete the P&R process. The process was divided into 6 simplified steps and the median and range of days needed for each phase were estimated based on data reported in official/published documents. The analysis was stratified considering every single step of the assessment phase and included segmentation of drugs into indications for rare diseases, Orphan designation, Innovation assessment and Managed entry agreements (MEAs).
Results: Overall, 181 first indication procedures were submitted to AIFA in the period considered and, of these, 167 (92.3%) were completed and 129 procedures were considered for the final analysis and the median duration of the entire process (MAH submission to final Gazette publication) was 434 days (range 176.0-918.0). The duration of procedures for rare diseases (n = 53) was longer than those for non-rare-disease procedures (n = 76) (463.0 days vs 407.5 days respectively). Among rare disease procedures, orphan designation and MEAs represent predictors for time prolongation while innovation is associated with a shorter assessment time.
Conclusion: The study describes the time spent in each phase of the assessment and the appraisal process and demonstrates that uncertainty represents the main driver for the increment in the overall time.
Keywords: AIFA, CPR, CTS, Rare diseases, Negotiation times
Received: May 25, 2023
Accepted: September 26, 2023
Published online: October 31, 2023
Global & Regional Health Technology Assessment - ISSN 2283-5733 - www.aboutscience.eu/grhta
© 2023 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0).
Commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Introduzione
Negli ultimi anni, il progresso scientifico e tecnologico ha prodotto terapie sempre più complesse e a valore aggiunto importante. Questo ha generato un miglioramento della qualità di vita dei pazienti grazie a un effetto notevole sulla storia della malattia dei pazienti. Una conseguenza diretta di queste innovazioni ha come effetto la significativa pressione che sia i pazienti che i Sistemi Sanitari Nazionali (SSN) rivolgono a un accesso tempestivo alle nuove terapie.
Esiste una diffusa letteratura che ha effettuato analisi comparative in riferimento ai tempi di accesso alle terapie in Europa e in altri Paesi. Tuttavia, i risultati conclusivi in termini di rapidità della messa a disposizione dei farmaci e del loro impatto sociale ed economico sono difficili da trarre in virtù delle differenze sostanziali nei sistemi normativi e sanitari (1,2,3).
Nello specifico contesto dell’Unione Europea (UE), la maggior parte dei nuovi farmaci (e dei loro generici e biosimilari) viene autorizzata tramite una procedura centralizzata, gestita dall’Agenzia Europea dei Medicinali (EMA) e seguita da una decisione della Commissione Europea (CE). Questa procedura consente ai richiedenti di ottenere un’autorizzazione all’immissione in commercio valida in tutta l’UE e nei Paesi dello Spazio economico europeo (Islanda, Liechtenstein e Norvegia). Una volta concessa l’autorizzazione all’immissione in commercio dell’UE, il farmaco può in linea di principio essere commercializzato, anche se l’accesso dei pazienti ai nuovi medicinali è limitato dallo status di rimborsabilità in ciascun Paese membro. Infatti, la procedura di Prezzo e Rimborso (P&R) rimane una competenza nazionale e regionale e i suoi processi e conseguenti tempi variano profondamente da Paese a Paese. Alcune analisi hanno stimato che il tempo medio di approvazione in Europa tra la decisione della Commissione Europea e la messa a disposizione dei farmaci ai pazienti è pari a 511 giorni con una forte variabilità da Paese a Paese (min: 133 giorni in Germania; max: 899 giorni in Romania) (4).
In Italia, l’Agenzia Italiana del Farmaco (AIFA) ha pubblicato diverse analisi relative alle tempistiche delle procedure di Prezzo e Rimborso dei farmaci considerando in particolare il periodo 2018-2022 (5,6,7). Gli obiettivi di questi documenti erano quelli di garantire i principi di trasparenza della pubblica amministrazione, mostrando i tempi delle procedure di P&R condotte dall’AIFA nel periodo in analisi, e di informare i cittadini sui tempi di accesso dei pazienti ai farmaci rimborsati, sulla durata delle procedure di P&R e se questa fosse stata influenzata dalla pandemia di SARS-CoV-2 (Covid-19). Nell’ultimo report, pubblicato ad aprile 2023, l’Agenzia ha esaminato oltre 4.122 procedure tra il 2018 e il 2022 e ha stimato una durata media del processo di P&R (da approvazione CE alla pubblicazione in Gazzetta) pari a circa 327 giorni per i farmaci non generici e a 145 per i farmaci generici.
Altri studi a livello nazionale hanno cercato di analizzare le specifiche tempistiche legate all’accesso regionale (8), alla comparazione tra le Commissioni che hanno gestito la fase di valutazione in periodi differenti (9) o al potenziale impatto che ha avuto la pandemia di SARS-CoV-2 (5). A oggi, a nostra conoscenza, poco è stato pubblicato relativamente ai farmaci orfani e alle determinanti delle tempistiche necessarie per l’approvazione e la messa a disposizione di questa particolare categoria di medicinali.
Recentemente, nel corso del 2022, il capitolo italiano (Italy Rome Chapter) dell’International Society for Health Economics and Outcomes Research (ISPOR), in collaborazione con PharmaLex Italia, ha condotto un progetto multistakeholder i cui obiettivi erano l’individuazione e l’analisi delle difficoltà che a oggi si riscontrano nella negoziazione di P&R in Italia sulle malattie rare (10). Il documento ha avuto come primo step metodologico proprio l’analisi delle tempistiche AIFA che, partendo da un database messo a disposizione da PharmaLex Italy S.p.A., aveva come finalità ultima quella di proporre soluzioni pratiche che potrebbero ottimizzare i processi di assessment e appraisal dei farmaci per malattie rare all’interno del percorso negoziale.
Il principale obiettivo di questo articolo è stato quello di aggiornare e sviluppare nel dettaglio l’analisi condotta dal gruppo dell’ISPOR Italy Rome Chapter riguardo alle tempistiche di negoziazione di P&R delle nuove molecole introdotte in Italia tra il 2018 e il 2022. In particolare, l’analisi ha effettuato un primo confronto delle tempistiche tra farmaci per malattie rare e non. Successivamente, all’interno dei farmaci per malattie rare, si è tentato di identificare le determinanti che influenzano i tempi di negoziazione AIFA per queste molecole.
Metodi
Il processo di Prezzo e Rimborso in Italia e definizione degli step temporali
Il processo di valutazione e negoziazione HTA è stato originariamente definito dal Comitato interministeriale per la pianificazione economica (CIPE), Risoluzione n. 3 del 1º febbraio 2001. Questa definizione rimane valida fino ad agosto 2020 quando un nuovo Decreto interministeriale stabilisce nuovi criteri per la determinazione dei prezzi e del rimborso da applicare entro il 1 marzo 2021 con nuove Linee Guida strutturate per lo sviluppo del dossier P&R (5,8,11).
Al fine di semplificare il processo in questa analisi sono stati definiti 6 step principali (Fig. 1):
– Step 1: dalla sottomissione del dossier di P&R all’apertura della procedura in Commissione Tecnico-Scientifica (CTS);
– Step 2: dall’apertura della procedura in CTS al parere della stessa Commissione;
– Step 3: dal parere della CTS all’apertura dell’istruttoria in Commissione Prezzo e Rimborso (CPR);
– Step 4: dall’apertura dell’istruttoria CPR al parere della stessa Commissione;
– Step 5: dal parere della CPR alla ratifica dell’accordo da parte del Consiglio di Amministrazione (CdA) di AIFA;
– Step 6: dalla ratifica del CdA alla pubblicazione in Gazzetta Ufficiale (GU).
Data source
Le informazioni utilizzate per la costruzione del dataset oggetto dell’analisi sono state reperite dalle principali fonti informative pubbliche:
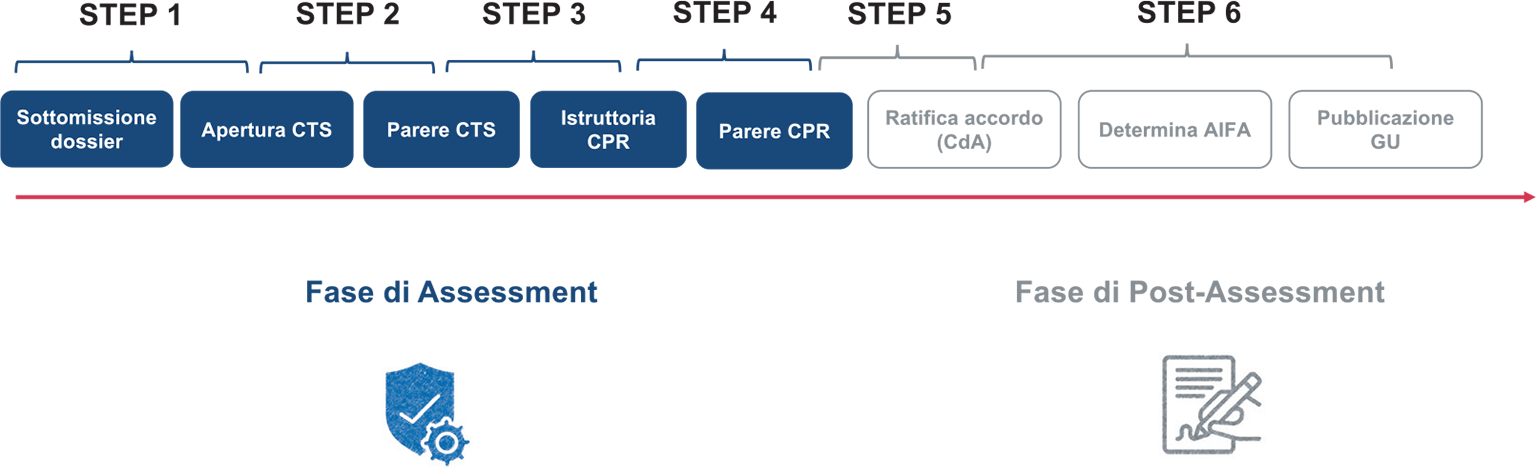
Fig. 1 - Step valutativi del proecesso di Prezzo e Rimborso.
– caratteristiche molecola: farmaco con indicazione per il trattamento di malattie rare (12), riconoscimento status di farmaco orfano EMA (13), riconoscimento status di farmaco innovativo AIFA (14,15), negoziazione di un Managed Entry Agreement (MEA) (14), presenza di uno sconto confidenziale (14);
– tempistiche AIFA: data della pubblicazione in GUUE (16), data di apertura e di chiusura (parere) del processo valutativo di CTS e CPR, data della determina AIFA, data della ratifica dell’accordo da parte del CdA, data di pubblicazione in GU, data di efficacia e validità della GU, indicazione rimborsata e classe di rimborsabilità del farmaco, collocazione del farmaco in classe Cnn, regime di fornitura del farmaco (14), data di tutte le riunioni effettuate da CTS e CPR (17).
– La raccolta di tali informazioni è stata centralizzata all’interno di un database realizzato ad hoc da PharmaLex S.p.A. Il database, costruito in ambiente Microsodt Excel® è stato popolato con le informazioni riguardanti i farmaci di nuova registrazione (prima indicazione di nuove entità chimiche/biotecnologiche) riportate negli esiti dell’Ufficio Procedure Centralizzate di AIFA.
Criteri di inclusione e stratificazione dei risultati
La selezione dei farmaci inclusi nell’analisi si è basata sulle procedure aperte dalla data del 28 settembre 2018 (elezione delle Commissioni Tecnico-Scientifica e Prezzo e Rimborso in carica al momento della stesura di questo paper) e che siano state chiuse entro la GU del 12.02.2023 (cut-off dell’analisi). Sono stati pertanto esclusi dall’analisi i farmaci con procedura in corso (n = 152), i farmaci che hanno iniziato la procedura con la vecchia Commissione (apertura CTS precedente a ottobre 2018 n = 153) e i farmaci con esito negativo di rimborsabilità (classe C, n = 38) (Fig. 2).
L’analisi sulle tempistiche nei vari step considerati è stata effettuata stratificando le procedure in relazione a:
– farmaci per Malattie non Rare (FMnR): include le molecole che non hanno un’indicazione per malattia rara come successivamente definito;
– farmaci per Malattie Rare (FMR): tutti i farmaci che abbiano un’indicazione per malattia rara definita da una prevalenza di non più di 5 soggetti ogni 10.000 individui;
– farmaci per Malattia Rara che hanno negoziato un MEA: farmaci per cui risulti negoziato un Managed Entry Agreement secondo quanto riportato in GU;
– farmaci Orfani per Malattie Rare (FOMR): Farmaci che rispettino i tre criteri per la definizione di farmaco orfano (Online) e presenti su database orphanet (Online):
⚬ devono essere indicati per una patologia che mette in pericolo la vita o debilitante in modo cronico
⚬ devono essere indicati per una condizione clinica rara, definita da una prevalenza di non più di 5 soggetti ogni 10 mila individui, calcolata a livello dell’Unione Europea
⚬ non devono essere disponibili trattamenti validi o, se sono già disponibili dei trattamenti, il nuovo farmaco deve rappresentare un beneficio clinico significativo;
– farmaci non Orfani per Malattie Rare (FnOMR): farmaci con indicazione per malattia rara ma senza il riconoscimento di orfanicità come definito nel punto precedente;
– farmaci Orfani per Malattie Rare Innovativi (FOMRI): riconoscimento da parte di AIFA del criterio di innovatività piena o condizionata secondo quanto riportato in GU;
– farmaci Orfani per Malattie Rare non Innovativi (FOMRnI): nessun riconoscimento da parte di AIFA del criterio di innovatività piena o condizionata.
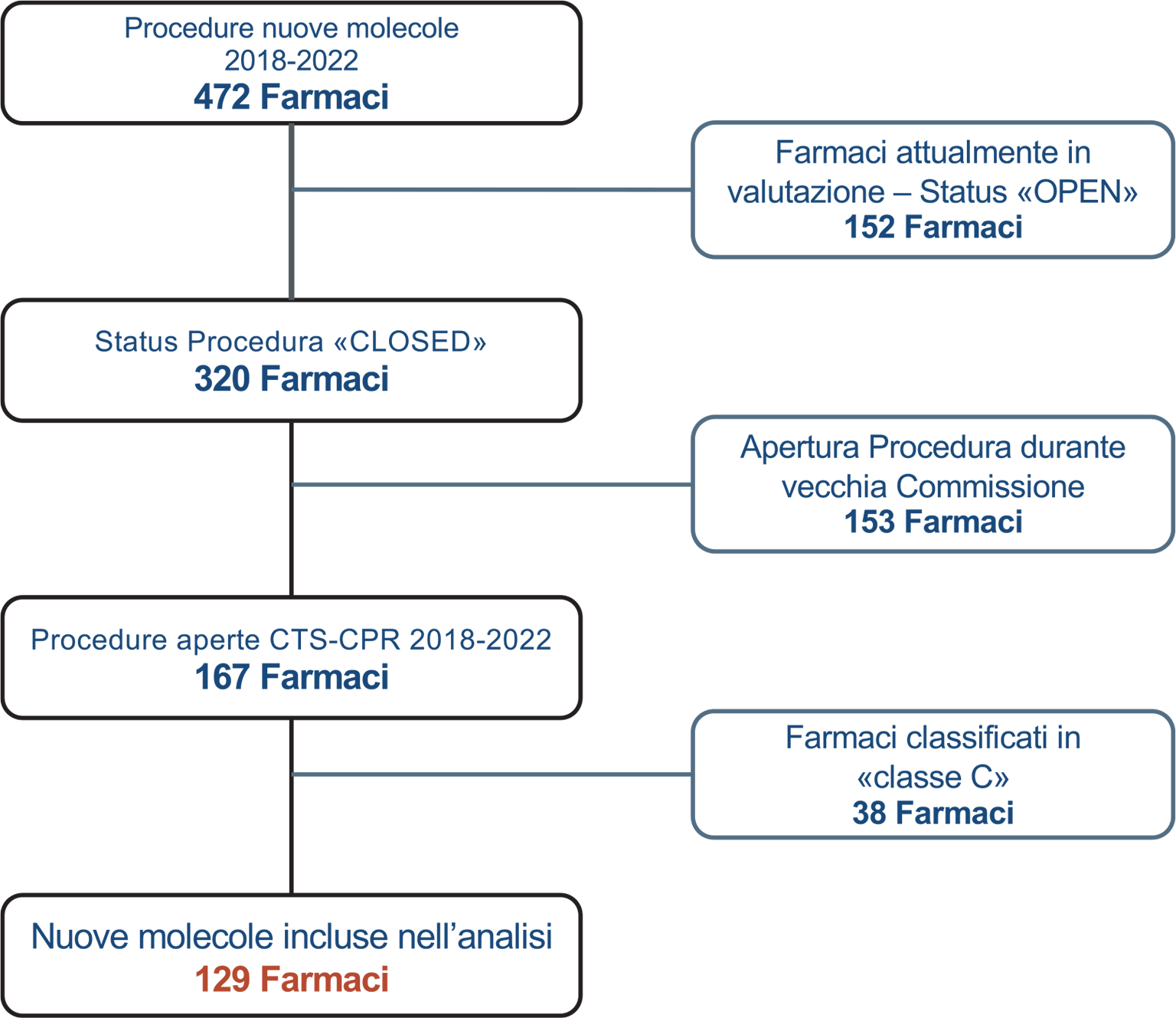
Fig. 2 - Flow-chart delle procedure incluse nell’analisi.
I diversi gruppi sono stati confrontati in termini di tempistiche in giorni per i differenti step considerati, funzioni di densità delle tempistiche per molecola, numero di rinvii, approfondimenti e audizioni CTS e CPR.
Risultati
L’analisi complessiva è stata condotta considerando 129 nuovi farmaci che hanno soddisfatto i criteri di inclusione nell’analisi. Di questi, 53 (58,9%) erano farmaci per malattia rara di cui 11 (8,5%) hanno negoziato un Managed Entry Agreement (MEA), mentre 43 (33,3%) hanno ottenuto la designazione di farmaco orfano. Dei 43 farmaci orfani, 26 (20,2%) erano farmaci a cui è stata riconosciuta l’innovatività terapeutica (Tab. I).
I 129 farmaci rimborsati hanno impiegato una mediana di 434,0 giorni dalla sottomissione del dossier di P&R alla pubblicazione della GU. La Figura 2 mostra in dettaglio i tempi mediani di tutti gli step della procedura negoziale. Complessivamente, i giorni trascorsi dalla sottomissione del dossier di P&R all’apertura della procedura in CTS sono stati 118,0 (31% del tempo complessivo), i giorni impiegati dalla CTS per esprimere il proprio parere sono stati 34,0 (9%) e 57,0 (15,1%) i giorni intercorsi dal parere della CTS all’apertura dell’istruttoria in CPR. I giorni impiegati dalla CPR per raggiungere l’accordo negoziale sono stati 91,0 (24%), mentre 36,0 (9,5%) sono stati i giorni necessari alla ratifica dell’accordo da parte del CdA. Infine, i giorni trascorsi dalla ratifica dell’accordo alla pubblicazione in GU sono stati 41,0 (10,9%).
Confrontando le tempistiche dei FMR vs FMnR (Fig. 2), si osserva che il tempo mediano totale di valutazione dei FMR risulta essere stato del 14% più lungo rispetto al tempo impiegato per i FMnR. Nello specifico, il tempo dei FMR è stato di 463,0 giorni (min 279,0 e max 877,0) e di 407,5 giorni (min 176,0 e max 918,0) per i FMnR.
Il tempo di permanenza nella CTS dei FMR è risultato quasi doppio rispetto ai FMnR (61,0 vs 31,5 [+29,5 giorni]), così come il tempo di permanenza in CPR (124,0 vs 64,0 [+60,0 giorni]).
Declinando i FMR secondo la caratteristica di orfano/non orfano (FOMR vs FnOMR), si nota come lo status di orfano rappresenti un fattore importante nell’allungamento dei tempi in CTS (3 volte più lungo) (63,0 vs 15,5 [+47,5 giorni]) e in CPR (2,4 volte più lungo) (140,0 vs 41,5 [+98,5 giorni]). Il tempo totale dei FOMR è, però, solo del 20% più lungo rispetto ai FnOMR (474,0 vs 396,5 [+77,5 giorni]) e questo può essere spiegato da una significativa riduzione di tutti gli altri momenti valutativi; in particolare si segnala, per i FOMR, la riduzione del periodo che intercorre tra il deposito e l’apertura in CTS (−20%) (99,0 vs 124,0 [−25,0 giorni]) (Fig. 3a).
| Nuove molecole | N | Durata processo complessivo in giorni
Mediana (min; max) |
Durata processo complessivo in giorni
Media (Ds) |
p-value* |
|---|---|---|---|---|
| Overall | 129 | 434,0 (176,0; 918,0) | 467,5 (157,2) | |
| Farmaci con indicazione per malattia rara (N Tot = 129) | ||||
| No (FMnR) | 76 (58,9%) | 407,5 (176,0; 918,0) | 462,7 (168,5) | |
| Sì (FMR) | 53 (41,1%) | 463,0 (279,0; 877,0) | 474,2 (140,8) | 0,330 |
| Sottogruppo nelle sole malattie rare (N Tot = 53) | ||||
| 1. Presenza di un MEA negoziato per malattie rare | ||||
| No | 42 (32,6%) | 462,0 (273,0; 877,0) | 468,4 (129,3) | |
| Sì | 11 (8,5%) | 486,0 (270,0; 866,0) | 496,6 (184,2) | 0,830 |
| 2. Riconoscimento dello status di farmaco orfano | ||||
| No (FnOMR) | 10 (7,8%) | 396,5 (273,0; 660,0) | 431,1 (112,7) | |
| Sì (FOMR) | 43 (33,3%) | 474,0 (279,0; 877,0) | 484,3 (145,9) | 0,280 |
| Sottogruppo nei soli farmaci orfani per malattie rare (N Tot = 43) | ||||
| 2.1 Riconoscimento dell’innovatività | ||||
| No (FOMRnI) | 17 (13,2%) | 496,0 (297,0; 877,0) | 532,6 (169,9) | |
| Sì (FOMRI) | 26 (20,2%) | 443,5 (270,0;748,0) | 452,7 (121,1) | 0,120 |
MEA: Managed Entry Agreement
Ds: Deviazione standard
*Non parametric Wilcoxon Rank Sum Test
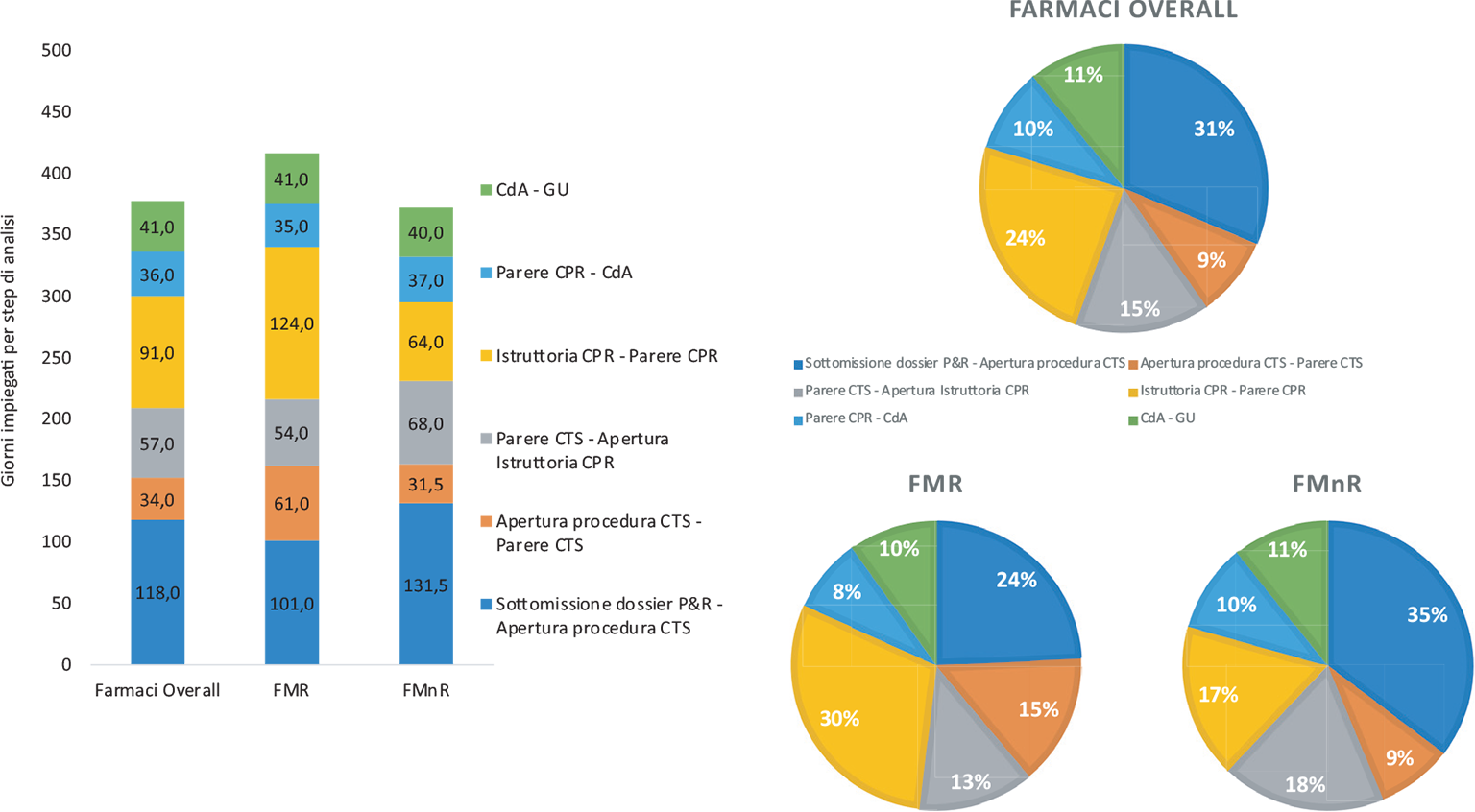
Fig. 3 - Valori mediani dei giorni per step di analisi, distribuzione percentuale della timeline complessiva (n = 129 nuove molecole) e distribuzione sottogruppo dei farmaci per malattie rare (FMR, n = 53) e non rare (FMnR, n = 76) (periodo di riferimento: 2018-2023).
Nel campione preso in esame, dei 53 FMR, quelli ai quali è stato accordato un MEA sono stati 11.
Il tempo mediano totale è stato di 486,0 giorni, +24 giorni rispetto ai 42 FMR che non hanno negoziato un MEA. La differenza più evidente è stata riscontrata nelle tempistiche di permanenza in CTS che risultano essere quasi 2 volte più lunghe nei FMR con MEA (mediana CTS 127,0 vs 44,5 [+82,5 giorni]). Il tempo mediano della permanenza in CPR risulta invece uguale (mediana CPR 124,0 vs 124,5 [0,5 giorni]) (Fig. 3a).
L’attribuzione dell’innovatività terapeutica ai FOMR (Fig. 3b) riduce leggermente il tempo totale rispetto alla mediana dei FOMR (mediana 443,0 vs 474,0 [−31,0 giorni]) ma non modifica il tempo di permanenza in CTS (76,5 vs 63,0 giorni). Si nota invece una riduzione della permanenza in CPR (126,0 vs 140,0). La mancanza del requisito di innovatività (FOMRnI) non modifica significativamente il tempo di permanenza in CTS (mediana 52,0 giorni); tuttavia, un tempo di permanenza in CPR di circa 5 mesi rende il tempo totale di valutazione il più lungo tra quelli valutati (496,0 giorni).
• Il tempo di permanenza nella CTS dei FOMRI è risultato più lungo del 47% rispetto ai FOMRnI (76,5 vs 52,0 [+24,5 giorni]).
• Il tempo di permanenza in CPR dei FOMRI è risultato più breve del 19% rispetto ai FOMRnI (126,0 vs 156,0 [−30,0 giorni]).
La Figura 5 mostra la densità delle distribuzioni dei giorni totali del processo di valutazione e negoziazione dei farmaci presi in esame sia come overall che declinati nei sottogruppi analizzati.
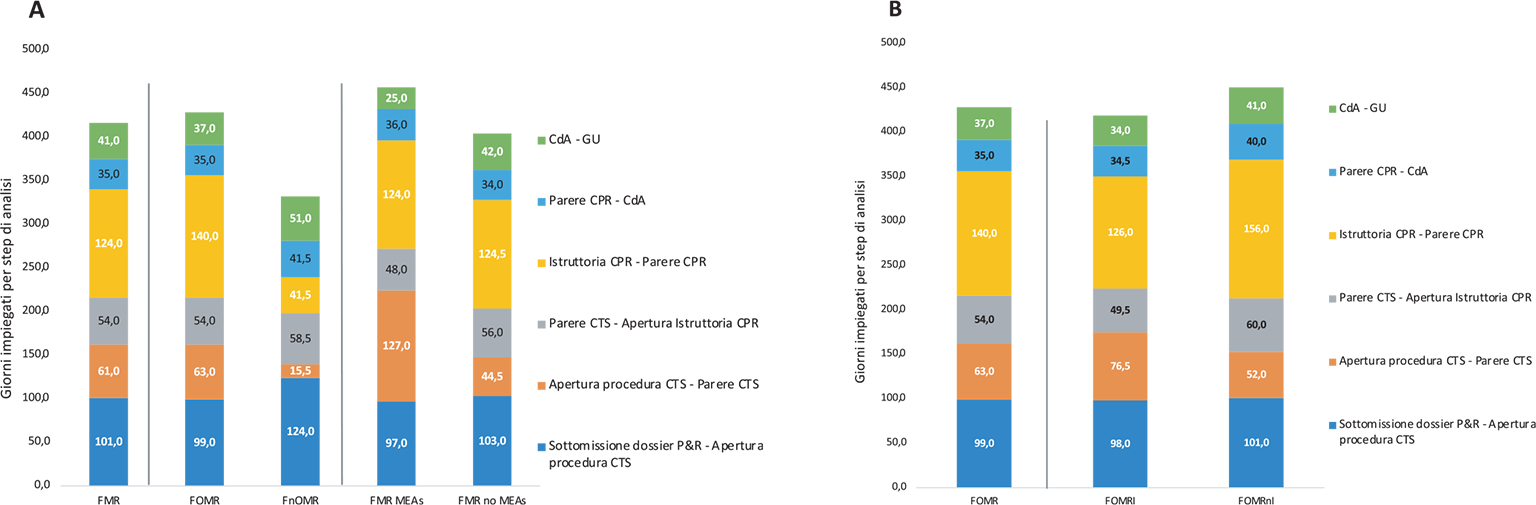
Fig. 4 - Valori mediani dei giorni per step di analisi per malattie rare (n = 53) e sottogruppi: a) farmaci per malattie rare orfani (FOMR, n = 43) e non orfani (FnOMR, n = 10); farmaci per malattie rare che hanno negoziato un MEA (FMR MEA, n = 11) e che non hanno negoziato un MEA (FMR no MEA, n = 42); b) farmaci per malattie rare orfani innovativi (FOMRI, n = 26) e non innovativi (FOMRnI, n = 17) (periodo di riferimento: 2018-2023).
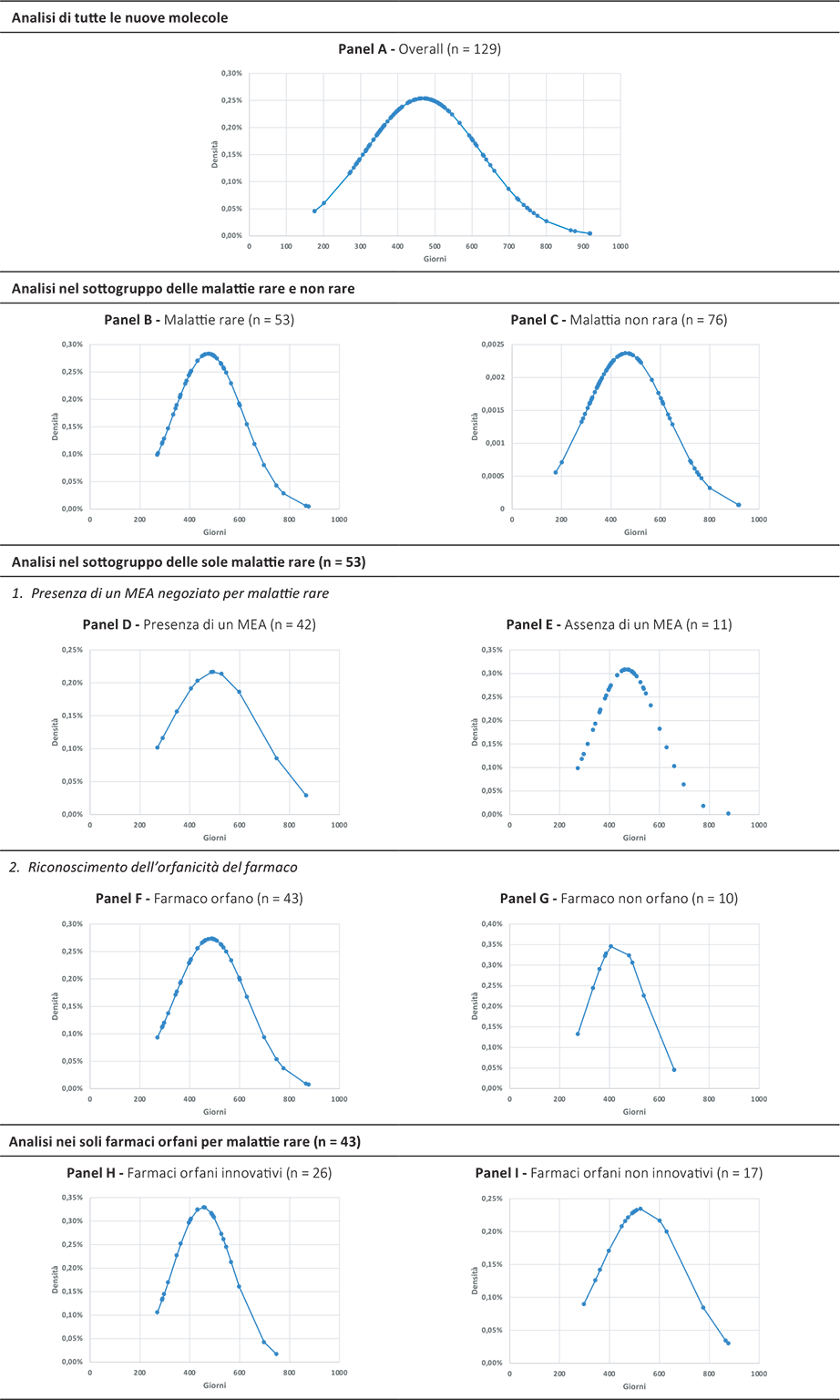
Fig. 5 - Densità delle distribuzioni dei giorni totali del processo di valutazione e negoziazione delle 129 nuove molecole incluse nelle analisi e nei sottogruppi di interesse (periodo di riferimento: 2018-2023).
Come si evince nessuno dei farmaci considerati ha chiuso il processo regolatorio di P&R sotto i 200 giorni complessivi.
La Tabella II, oltre a riproporre la mediana dei giorni totali del processo valutativo e dei giorni di permanenza in CTS e CPR dei farmaci presi in esame, mostra anche il numero mediano di rinvii, approfondimenti e audizioni avvenuti in CTS, nonché di rinvii, approfondimenti e convocazioni avvenuti in CPR. Dall’analisi si evince come i FMR richiedano più passaggi sia in CTS che in CPR rispetto ai FMnR, evidenziando al contempo come il driver principale sia lo status di “farmaco orfano”. Infatti, i FnOMR hanno un percorso decisamente più veloce.
Anche la negoziazione di un MEA rende il percorso di questi farmaci più lungo, confermato da un numero maggiore di approfondimenti sia in CTS che in CPR.
Il non riconoscimento dell’innovatività terapeutica dei farmaci orfani, nonostante sembri allungare di molto i tempi di valutazione, soprattutto in CPR, sembra non essere collegato a un numero maggiore di rinvii, approfondimenti o convocazioni.
Discussione
Questo lavoro ha tentato di quantificare le tempistiche di negoziazione di P&R delle nuove molecole ammesse a rimborso e quindi introdotte in Italia tra il 2018 e il 2022, sottolineando le principali caratteristiche che contraddistinguono i farmaci per le malattie rare. L’analisi ha mostrato come i FMR determinino un allungamento delle tempistiche regolatorie di quasi due mesi rispetto ai FMnR. Questo nonostante gli Uffici Tecnici di AIFA sembrino velocizzare gli step amministrativi riducendo i tempi di apertura del processo in CTS dal momento della sottomissione del dossier (circa 30,5 giorni in meno per FMR vs FMnR) e nella fase di passaggio tra parere CTS e apertura CPR (−14 giorni). Infatti, il notevole ritardo legato al processo per i FMR rispetto alle altre valutazioni è fortemente legato alla fase di appraisal della CTS (mediana 61 vs 31,5 giorni rispettivamente per FMR e FMnR) e della CPR (mediana 124 vs 64 giorni rispettivamente per FMR e FMnR).
| Numero di giorni
Mediana (min; max) |
CTS
Mediana (min; max) |
CPR
Mediana (min; max) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tot | CTS | CPR | Rinvii | Approfondimenti | Audizioni | Rinvii | Approfondimenti | Convocazioni | ||
| Overall (n = 129) | 434 (176; 918) | 34 (1; 577) | 91 (1; 542) | 0 (0; 3) | 2 (0; 8) | 0 (0; 2) | 0 (0; 3) | 2 (0; 10) | 0 (0; 4) | |
| FMnR - Malattia non rara (n = 76) | 407,5 (176; 918) | 31,5 (1; 577) | 64 (1; 542) | 0,5 (0; 3) | 1 (0; 8) | 0 (0; 2) | 0 (0; 3) | 2 (0; 10) | 0 (0; 2) | |
| FMR - Malattia rara (n = 53) | 463 (279; 877) | 61 (1; 371) | 124 (1; 479) | 0 (0; 3) | 2 (0; 6) | 0 (0; 1) | 0 (0; 2) | 3 (0; 7) | 0 (0; 4) | |
| P-value | 0,33 | 0,21 | 0,006 | 0,13 | 0,41 | 0,98 | 0,41 | 0,34 | 0,02 | |
| Sottogruppo nelle sole malattie rare (N Tot = 53) | ||||||||||
| 1. Presenza di un MEA negoziato per malattie rare | ||||||||||
| No (n = 42) | 462 (273; 377) | 44,5 (1; 371) | 124,5 (1; 457) | 0 (0; 3) | 2 (0; 5) | 0 (0; 1) | 0 (0; 2) | 3 (0; 6) | 0 (0; 4) | |
| Sì (n = 11) | 486 (270; 866) | 127 (1; 287) | 124 (52; 479) | 0 (0; 2) | 3 (1; 6) | 0 (0; 1) | 0 (0; 1) | 4 (1; 7) | 1 (0; 2) | |
| P-value | 0,83 | 0,01 | 0,41 | 0,40 | 0,07 | 0,95 | 0,98 | 0,25 | 0,008 | |
| 2. Riconoscimento dell’orfanicità del farmaco | ||||||||||
| FnOMR - No (n = 10) | 396,5 (273; 660) | 15,5 (1; 205) | 41,5 (1; 287) | 0 (0; 2) | 0,5 (0; 3) | 0 (0; 1) | 0 (0; 1) | 2,5 (0; 5) | 0 (0; 1) | |
| FOMR - Sì (n = 43) | 474 (270; 877) | 63 (1; 371) | 140 (1; 479) | 0 (0; 3) | 2 (0; 6) | 0 (0; 1) | 0 (0; 2) | 3 (1; 7) | 0 (0; 4) | |
| P-value | 0,28 | 0,22 | 0,007 | 0,91 | 0,01 | 0,92 | 0,70 | 0,41 | 0,24 | |
| Sottogruppo nei soli farmaci orfani per malattie rare (N Tot = 43) | ||||||||||
| 2 Riconoscimento dell’innovatività | ||||||||||
| FOMRnI - No (n = 17) | 496 (297; 877) | 52 (1; 212) | 156 (1; 479) | 0 (0; 3) | 2 (0; 5) | 0 (0; 1) | 0 (0; 2) | 3 (1; 7) | 0 (0; 4) | |
| FOMRI - Sì (n = 26) | 443,5 (270; 748) | 76,5 (1; 371) | 126 (1; 373) | 0 (0; 3) | 2 (1; 6) | 0 (0; 1) | 0 (0; 2) | 3 (1; 6) | 1 (0; 2) | |
| P-value | 0,12 | 0,60 | 0,43 | 0,89 | 0,25 | 0,90 | 0,41 | 0,56 | 0,11 | |
Analizzando nel dettaglio le motivazioni di questo ritardo, l’analisi ha mostrato come uno dei fattori determinanti sia rappresentato dal numero di approfondimenti richiesti da parte delle Commissioni (maggiore sia in CPR che in CTS per FMR rispetto a FMnR). Questo suggerisce che l’incertezza legata alla definizione di efficacia e valore economico di tali farmaci possa generare una maggiore necessità di approfondimento dei dati a disposizione ampliando, di conseguenza, i tempi di accordo tra i due attori della negoziazione. Tale effetto sembrerebbe essere confermato anche dall’importante differenza nei tempi di negoziazione nel momento in cui consideriamo altri parametri che rendono la definizione del valore di un farmaco incerta. Nel momento in cui confrontiamo le tempistiche in caso di un farmaco orfano, i tempi di permanenza in AIFA salgono a 486 giorni rispetto ai 396,5 dei farmaci non orfani per malattie rare. Anche la presenza di un MEA concordato nella negoziazione, solitamente utilizzato come strumento di condivisione dell’incertezza, è associato a un incremento di oltre 20 giorni per la conclusione del processo negoziale. Viceversa, laddove le prove sono robuste e l’efficacia incrementale è importante (parametri di innovazione di AIFA), il numero di giorni del processo dei farmaci orfani per malattie rare si riduce di oltre 50 giorni (mediana 443,5 vs 496,0 per FOMRI vs FOMRnI).
Esaminando il comportamento delle Commissioni, la CTS impiega una mediana di 34 giorni per esprimere un parere per le nuove entità chimiche approvate in Italia. Ne risultano maggiormente sfavoriti i farmaci orfani (mediana 63 giorni per FOMR vs 15,5 giorni per FnOMR) e i farmaci approvati con accordo di un MEA (127 giorni per FMR MEA vs 44,5 giorni per FMR no MEA). Il riconoscimento dell’innovatività per i farmaci per malattia rara orfani si associa a un incremento dei tempi mediani di valutazione della CTS di circa 20 giorni. Viceversa, la CPR in presenza di farmaci innovativi riduce le tempistiche di 30 giorni raggiungendo l’accordo di prezzo con l’azienda in tempi ristretti rispetto ai farmaci non innovativi. Tuttavia, i tempi di negoziazione del prezzo sono in generale notevolmente più alti rispetto ai tempi della CTS (mediana di 91 giorni rispetto ai 34 giorni della CTS). La maggiore durata del percorso valutativo in CPR si evidenzia principalmente rispetto allo status di farmaco orfano per malattia rara, la cui permanenza in CPR è superiore di oltre 100 giorni rispetto ai FnOMR (mediana 140 giorni vs 44,5). Questo, ancora una volta, potrebbe essere attribuito a un elevato livello di incertezza legato a prezzi dei comparatori che possano ancorare la negoziazione tra regolatore e azienda.
I risultati della nostra analisi sono in linea rispetto a quanto disponibile in letteratura anche se con alcune interessanti peculiarità. I report pubblicati da AIFA sui tempi di negoziazione e permanenza dei dossier di P&R relativi al periodo 2018-2022 (5,6,7) stimano una durata mediana dell’intero procedimento (definita come tempo impiegato dal completamento della verifica amministrativa alla data di conclusione dell’iter valutativo di CTS e CPR) per i FOMR pari a 284,5 giorni. Se osserviamo gli step 2-4 (Apertura CTS – Parere CPR) della nostra analisi, i dati sono sostanzialmente sovrapponibili avendo registrato una mediana di 310 giorni. Le differenze sono da attribuire ai criteri di inclusione considerati e la nostra analisi considera le sole procedure aperte e chiuse con accordo di rimborsabilità per il SSN nel periodo 2018-2022, mentre il report AIFA includeva tutte le procedure aperte. Inoltre, gli step considerati all’interno del processo negoziale non avevano la medesima definizione.
Se confrontiamo i risultati di questa analisi con quanto pubblicato negli anni passati con approcci simili (9), i dati mostrano un sostanziale aumento dei tempi impiegati tra l’apertura CTS e la determina in GU. Questa dilatazione dei tempi si è verificata sia rispetto ai tempi impiegati dalla stessa Commissione nel periodo 2018-2020 (durata media 287 giorni) sia rispetto alla Commissione precedente in carica nel periodo 2015-2018 (durata media 242 giorni vs 341 giorni nell’analisi attuale per l’anno 2022 (Tabella Supplementare A) considerando di escludere 118 giorni mediani per apertura CTS per rendere paragonabile questo valore con il medesimo periodo degli anni precedenti pubblicati) (9). Questo risulta evidente anche nella nostra analisi considerando l’andamento annuo delle tempistiche AIFA su tutte le categorie iniziali. La mediana dell’intero processo negoziale per il campione analizzato è passata da 346,5 giorni per il 2019 a 459,5 giorni nel 2022 (+33%). Questo è ancora più vero per i FMR (durata mediana 334,0 giorni nel 2019 vs 468,5 giorni nel 2022 (+40,3%) (Tabella Supplementare A).
Un’analisi con criteri simili a questo lavoro è stata presentata nel Rapporto annuale dell’Osservatorio Farmaci Orfani (OSSFOR) (18). Gli Autori si focalizzano sui soli farmaci orfani e stimano tempistiche di accesso EMA e AIFA. Per lo specifico step negoziale nazionale, il rapporto analizza un campione di 135 farmaci autorizzati da EMA tra Dicembre 2017 e Dicembre 2021. In questo caso si stima un tempo medio tra autorizzazione EMA e pubblicazione in Gazzetta di 19 mesi (mediana 15 mesi; Min: 2 mesi, Max: 92 mesi). Considerando l’approssimazione in mesi del rapporto OSSFOR possiamo considerare il dato sostanzialmente sovrapponibile ai nostri risultati, con la differenza che il nostro lavoro ha considerato come step di start il momento della sottomissione del dossier in AIFA, mentre il lavoro dell’Osservatorio ha considerato l’approvazione EMA. Inoltre, il campione del rapporto OSSFOR era maggiormente numeroso in quanto considerava tutte le indicazioni registrate in EMA per farmaci orfani mentre il nostro lavoro si è focalizzato sulle sole nuove entità chimiche limitando i bias legati alle estensioni di indicazione.
Rispetto alla letteratura disponibile, questa analisi consente di ottenere un focus molto dettagliato sui tempi negoziali in Italia riguardo alle nuove molecole indicate per le malattie rare. Le particolarità di accesso di queste tecnologie risiedono spesso nell’incertezza legata alla definizione e alla misurazione dei parametri di efficacia e safety rispetto a uno standard of care, molto spesso di altrettanto difficile definizione. Questo problema è strettamente legato alle caratteristiche degli studi clinici impiegati per validare le prove di efficacia per i quali spesso vengono evidenziati limiti di arruolamento e rappresentatività delle coorti arruolate particolarmente carenti nel caso di farmaci orfani e malattie rare (19). Di conseguenza, nell’esaminare i tempi di accesso dei farmaci per le malattie rare si riscontra un effetto controintuitivo per cui, nonostante il bisogno terapeutico maggiore, i tempi con cui la tecnologia può raggiungere i pazienti si allungano. Gli effetti del percorso regolatorio sono rappresentati dal maggior numero di approfondimenti richiesti dalle Commissioni AIFA per le malattie rare e/o i farmaci orfani rispetto alle altre molecole incluse nella nostra analisi.
Un importante limite di questa analisi è sicuramente la sola visione nazionale delle tempistiche regolatorie che rende incompleta la traiettoria nazionale dei farmaci per le malattie rare dall’autorizzazione EMA alla messa a disposizione della tecnologia al paziente. Infatti, a oggi non è disponibile una stima analitica aggiornata dei tempi di accesso regionale per approvazione e dispensazione dei farmaci a livello locale dopo la pubblicazione in GU. Lo studio di Russo et al. del 2010 (8), ha utilizzato i dati di tracciabilità del Ministero della salute per identificare i tempi di prima movimentazione dei farmaci oncologici autorizzati tra il 2006 e il 2008. Le Regioni impiegavano una media di 160 giorni per effettuare la prima movimentazione dalla chiusura del processo AIFA registrando un range particolarmente elevato che andava da un minimo di 40 a un massimo di 540 giorni dalla pubblicazione in GU. Più recentemente, il Rapporto annuale dell’Osservatorio Farmaci Orfani (OSSFOR) (19), che pubblica annualmente un monitoraggio delle tempistiche di prima movimentazione dei farmaci orfani nelle Regioni italiane, stima delle tempistiche che variano da un minimo di 1.638 giorni prima della GU a un massimo di oltre 1.762 giorni dopo la pubblicazione in GU. Tali dati, anche se legati a un periodo storico differente e a criteri di inclusione diversi dalla nostra analisi e con una serie di limitazioni associate alla definizione di approvazione regionale (la movimentazione non vuol dire approvazione nei prontuari terapeutici), spingono i decisori a una riflessione e a un’attenta analisi circa gli step e le metodiche approvative che ciascuna Regione mette in atto una volta definiti il prezzo e la rimborsabilità a livello nazionale.
Un secondo limite dello studio risiede in possibili bias delle tempistiche legate al periodo storico analizzato. I dati raccolti fanno riferimento al periodo a cavallo della pandemia per cui è verosimile che alcuni ritardi siano da attribuire alle difficoltà legate, principalmente a inizio 2020, all’adattamento delle Commissioni e degli Uffici Tecnici di AIFA a un nuovo metodo di lavoro a distanza oltre che alla prioritizzazione verso specifici farmaci per la lotta alla pandemia. Tuttavia, la natura comparativa dello studio (FMR vs FMnR) riferito al medesimo periodo e alcune analisi pubblicate da AIFA (6) sembrerebbero mitigare questo effetto.
Infine, sono da considerare la natura descrittiva dell’analisi e la limitazione della numerosità campionaria che non hanno consentito un’analisi statistica approfondita dei predittori delle tempistiche e un’adeguata stratificazione dei parametri da testare. Inoltre, non è stato possibile reperire informazioni specifiche sulle tempistiche delle 152 procedure escluse dall’analisi perché ancora in fase negoziale. Per tali procedure, infatti, non è stato possibile determinare la data di sottomissione, la classificazione e il tempo di chiusura del procedimento, dato che tali variabili vengono raccolte direttamente dalla pubblicazione in Gazzetta del prodotto. Questo potrebbe rappresentare un bias di selezione del campione e i risultati potrebbero cambiare in futuro.
Conclusioni
In conclusione, la nostra analisi evidenzia la necessità di definire in maniera chiara ed esaustiva le metodologie necessarie per la valutazione del valore dei farmaci per le malattie rare in modo da velocizzare i processi regolatori e da garantire ai pazienti l’accesso alle tecnologie innovative. Futuri lavori dovrebbero incentrarsi anche su un confronto internazionale sui tempi di tali processi tentando però di ancorare step temporali omogenei tra i differenti processi regolatori dei diversi Paesi (1,3).
La nuova governance di AIFA è chiamata a una missione importante che coniughi sostenibilità e tempi di accesso adeguati tentando di innovare il processo di Prezzo e Rimborso. Questo, ovviamente, dovrebbe essere l’obiettivo primario anche delle Regioni e la necessità di conoscere i tempi di accesso regionali può essere considerata come una delle finalità del prossimo futuro in modo da poter garantire un accesso rapido e uniforme su tutto il territorio nazionale.
Disclosures
Conflict of interest: AM, CG e PC declare they have no conflict of interest related to this article. PR is an employee of PharmaLex Italy S.p.A.
Financial support: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Bibliografia
- 1. Kwon HY, Kim H, Godman B. Availability and Affordability of Drugs With a Conditional Approval by the European Medicines Agency; Comparison of Korea With Other Countries and the Implications. Front Pharmacol. 2018;9:938. CrossRef PubMed
- 2. Oye KA, Eichler HG, Hoos A, Mori Y, Mullin TM, Pearson M. Pharmaceutical licensing and reimbursement in the European Union, United States, and Japan. Clin Pharmacol Ther. 2016;100(6):626-632. CrossRef PubMed
- 3. Zamora B, Maignen F, O’Neill P, Mestre-Ferrandiz J, Garau M. Comparing access to orphan medicinal products in Europe. Orphanet J Rare Dis. 2019;14(1):95. CrossRef PubMed
- 4. EFPIA Patients W.A.I.T. Indicator 2021 Survey. July 2022. Online (Accessed March 2023).
- 5. Gallo V, Alessi E, Montilla S, Altamura G, Traversa G, Trotta F. The timelines for the price and reimbursement authorization in Italy 2018-2020. Front Med (Lausanne). 2022;9:1055359. CrossRef PubMed
- 6. Strategia A, ed. Economia del Farmaco Settore Hta ed Economia Del Farmaco, Rapporto sulle tempistiche delle procedure di prezzo e rimborso dei farmaci nel quadriennio 2018-2022. Aprile 2022 – Online (accessed May 2022).
- 7. Farmaco ASEFSHEd. Rapporto sulle tempistiche delle procedure di prezzo e rimborso dei farmaci nel quadriennio 2018-2020. Settembre 2021. Online (Accessed March 2023)
- 8. Russo P, Mennini FS, Siviero PD, Rasi G. Time to market and patient access to new oncology products in Italy: a multistep pathway from European context to regional health care providers. Ann Oncol. 2010;21(10):2081-2087. CrossRef PubMed
- 9. Raimondo P, Casilli G, Isernia M, et al. [Not Available]. Glob Reg Health Technol Assess. 2020;7:109-114. CrossRef PubMed
- 10. Canonico, P.L., et al. Rare Disease Deep Dive & Proposals (EXPLORARE). Febbraio 2023. Online (Accessed March 2023).
- 11. AIFA. Decreto 2 agosto 2019. Criteri e modalita’ con cui l’Agenzia italiana del farmaco determina, mediante negoziazione, i prezzi dei farmaci rimborsati dal Servizio sanitario nazionale (20A03810) (GU Serie Generale n.185 del 24-07-2020), p.6. Online (Accessed February 2023).
- 12. Orphanet Report Series. Lists of medicinal products for rare diseases in Europe. Online (Accessed March 2023).
- 13. European Medicines Agency (EMA). Official website of the European Union. Human Medicines search. Online (Accessed March 2023).
- 14. Gazzetta ufficiale della Repubblica Italiana. Online (Accessed March 2023).
- 15. Agenzia Italiana del Farmaco (AIFA). Lista Farmaci Innovativi. Online (Accessed March 2023).
- 16. Gazzetta ufficiale dell’Unione europea. EUR-Lex Aaccesso al diritto dell’Unione Europea. Online (Accessed March 2023).
- 17. Agenzia Italiana del Farmaco (AIFA). Esiti delle riunioni CTS e CPR. Online (Accessed March 2023).
- 18. Osservatorio Farmaci Orfani (OSSFOR). VI Rapporto Annuale: Investimenti e trasparenza dei processi: le condizioni per garantire l’equità di accesso ai malati rari. 2022. Online (Accessed July 2023).
- 19. Cox GF. The art and science of choosing efficacy endpoints for rare disease clinical trials. Am J Med Genet A. 2018;176(4):759-772. CrossRef PubMed