|
Drug Target Insights 2025; 19: 11-17 ISSN 1177-3928 | DOI: 10.33393/dti.2025.3271 ORIGINAL RESEARCH ARTICLE |
Exploring the in vitro anti-diabetic potential and in silico studies of 2, 3 and 2, 6-dichloroIndolinone
Abdur Rauf![]() 1, Waqas Alam
1, Waqas Alam![]() 1, Momin Khan2, Hany W. Darwish3, Maria Daglia
1, Momin Khan2, Hany W. Darwish3, Maria Daglia![]() 4, Ahmed A. Elhenawy
4, Ahmed A. Elhenawy![]() 5, Haroon Khan
5, Haroon Khan![]() 1,6
1,6
ABSTRACT
Introduction: Adequate hyperglycemic control is still a huge challenge with the clinically used therapeutics. New, more effective anti-diabetic agents are on the top list of drug discovery projects.
Methods: This article deals with the in vitro anti-diabetic potential of 2, 3 dichloroIndolinone (C1) and 2, 6-dichloroIndolinone (C2) on α-glucosidase and α-amylase followed by in silico analysis.
Results: Both compounds, C-1 and C-2, caused significant inhibition of α-glucosidase at various test concentrations with IC50 of 35.266 μM and 38. 379 μM, respectively. Similarly, compounds C-1 and C-2 elicited significant anti-α-amylase action with IC50 values of 42.449 μM and 46.708 μM, respectively. The molecular docking investigation regarding the α-glucosidase and α-amylase binding site was implemented to attain better comprehension with respect to the pattern in which binding mechanics occur between the C1 and C2 molecules and the active sites, which illustrated a higher binding efficacy in appraisal with reference inhibitor and acarbose. The interactions between the active compounds C1 and C2 with the active site residues were mainly polar bonds, hydrogen bonding, π−π, and π−H interactions, which contributed to a strong alignment with the enzyme backbone. Similarly, effective binding is frequently indicated by a strong and stable hydrogen-bonding pattern, which is suggested by the minimal fluctuation in MM-PBSA values.
Conclusion: In short, this study will contribute to providing these compounds with an improved anti-diabetic profile and decreased toxicity.
Keywords: 2, 3 and 2, 6-dichloroIndolinone, α-glucosidase/α-amylase inhibition, Molecular docking, Molecular simulation
Received: August 23, 2024
Accepted: February 4, 2025
Published online: March 10, 2025
Corresponding author:
Haroon Khan
email: haroonkhan@awkum.edu.pk
Drug Target Insights - ISSN 1177-3928 - www.aboutscience.eu/dti
© 2025 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0).
Commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Introduction
Diabetes mellitus (DM) is a prolonged metabolic state that can result from reduced insulin synthesis or decreased insulin’s ability to facilitate the absorption of glucose. Hyperglycemia or extremely high blood glucose, and glucose intolerance are characteristics of DM (1). The World Health Organization (WHO) describes DM as a life-long metabolic disorder marked by increased blood glucose levels that eventually cause harm to the kidneys, eyes, nerves, cardiovascular system, and vascular system. Over ninety percent of the cases of DM are caused by type II DM (T2DM), which is described by tissue inadequate insulin or insulin resistance synthesis by the pancreatic β cells and an improper compensatory insulin secretory response (2). Diabetes is the tenth most common cause of death, accounting for over a million deaths annually. By 2030, the number of people with type 2 diabetes worldwide is expected to reach 7079 per 100,000, indicating a persistent increase in cases worldwide. Concerning evidence indicates that prevalence is on the rise in lower-income nations. There is a need for immediate clinical preventative and public health actions. (3). Type 2 diabetes, which develops in adult age, is defined by inadequate insulin production, either alone or in combination with insulin resistance (4). There are some key similarities between heart disease [cardiovascular disease (CVD)] and DM. One significant and unique risk factor for CVD is diabetes. The most frequent and expensive blood vessel consequence of diabetes is heart disease [coronary heart disease (CHD)] (5). There are presently no effective medications to treat this health condition, and the available therapies are successful for some people but not all. Innovative anti-diabetic drugs are therefore greatly needed. Diabetes management requires an extensive review of current pharmacotherapies and phytotherapies, as well as nutraceutical-based therapies for effective management of diabetes, along with their merits and limitations (6).
The first known oxindole derivative was discovered in the form of alkaloids from the bark of the tropical climber species known as cat claw’s plant (Uncaria tomentosa), which is native to the dense rainforests of the Amazon region and other tropical regions of central and southern South America (7). Oxindoles are naturally occurring aromatic chemical compounds found in a variety of plant materials as well as in animal tissues and body fluids. A six-membered benzene ring and a five-membered ring containing nitrogen combine to form an oxindole (8). Several researchers have discovered, produced, generated, and analyzed oxindole derivatives for a range of biological effects and observed significant effects (9-12). Medicinal chemists are motivated to create new oxindole derivatives because of the oxindole nucleus’s historical and current medicinal value. Oxindole-derived compounds are potent inhibitors of the α-glucosidase enzyme, which is a target enzyme in the treatment of DM (13). The breakdown and intestinal absorption of carbohydrates occur by important enzymes α-amylase and α-glucosidase, respectively. The therapy of non-insulin-dependent DM (NIDDM) can benefit greatly from the inhibition of these enzymes, which inhibits the rise in blood glucose levels following the consumption of carbohydrates (14). Similarly, 2, 3 and 2, 6-dichloroIndolinone have already shown potential drug-like properties coupled with antioxidant potential (15). In the present article, we are presenting the in vitro α-glucosidase and α-amylase activities of oxindoles, 2, 3 dichloroIndolinone (C1) and 2, 6-dichloroIndolinone (C2) (Figure 1) followed by molecular docking and molecular dynamic simulation.
Materials and Methods
Chemicals
Chemicals used were α-glucosidase, α-amylase, acarbose, glucopyranoside, phosphate buffer, and piperidine, Na2CO3, starch and dinitro salicylic acid -solution, hydrochloric acid and distilled water.
FIGURE 1 - Structures of test compounds.
α-Glucosidase Activity
In this test, Glucopyranoside is incorporated into the solvent (phosphate buffer). Several doses of the synthesized compounds, including 62.5 µg/mL, 125 µg/mL, 250 µg/mL, 500 µg/mL, and 1000 µg/mL, were used to create the sample solutions. Added 0.5 µg/mL of glucosidase in distilled water to the mixture described above. Subsequently, the resultant mixture was incubated at 37°C for 20 minutes (16). Hydrochloric acid was used to stop the reaction mixture after the incubation period. Using a spectrophotometer, the color intensity was determined at a wavelength of 540 nm. The percentage inhibition was computed using the following equation:
Inhibition (%) = (Control Abs − Test Abs/Control Abs) × 100
α-Amylase Activity
In this assay, previously published techniques were applied (16). α-amylase, phosphate buffer, and the sample solution were prepared by combining different amounts of the produced compounds (62.5 µg/mL, 125 µg/mL, 250 µg/mL, 500 µg/mL, and 1000 µg/mL). After mixing this solution with the starch solution, the mixture was left to incubate for around 20 minutes at 37°C. Following incubation, the reactant mixture was just maintained at 100°C in a water bath. At 656 nm, the color intensity was determined using a microplate reader. The following formula was used to get the inhibition percentage.
Inhibition (%) = (Control Abs - Test Abs/Control Abs) x 100
Molecular Docking
The synergistic effect of characteristics of active compounds C1 and C2 on α-glucosidase and α-amylase inhibitory actions was investigated using molecular docking studies (17). The protein data bank provided the α-glucosidase and α-amylase PDB IDs, which are 5NN5 and 4GQR, respectively.
Molecular Dynamic Simulation
We calculated the parameters, including binding free energy, van der Waals energy, electrostatic energy, kinetic energy, and potential energy changes using molecular dynamics simulations. Furthermore, we investigated the interactions that these compounds had with the proteins, glucosidase, and amylase throughout a time span of 0 to 250 ns. The binding free energy change values for the two compounds interacting with the proteins were also computed (18). The docking analysis was performed to explicate the potency of these desirable molecules in vitro against the epidermal growth factor receptor (EGFR) kinase through their potential interaction mechanisms with their crystal frameworks (PDB: 1b2y (19), PDB: 3A4A) (20) ). The docking investigation was implemented through Glide’s module®. The preliminary inhibitors (Acarbose) were relocked into the α-amylase crystal framework to verify the docking methodology. Furthermore, the efficacious performance of the targeted molecules was authenticated via the low values of RMSD (1.02 respectively), which were acquired through the root mean square deviation between the native and relocked poses of the co-crystallized inhibitor.
Statistical Analysis
The data is illustrated as mean ± SEM of three distinct measurements. Analyisis of variance (ANOVA) at *p < 0.05 and the Bonferroni test showed static significance using GraphPad version 8.
Results
Effects of Compounds on α-glucosidase Inhibition
Figure 2 shows the α-glucosidase inhibition of selected compounds (C1-C2) when studied at various concentrations. In the assay, C1 produced marked inhibition that was increased with increasing drug concentrations, and the maximum effect (88%) was noted at 1000 μM. The IC50 value of C1 was estimated as 35.266 μM. Similarly, C2 displayed significant α-glucosidase inhibition when tested at various concentrations. The maximum inhibitory effect was 85% at a dose of 1000 μM in specified conditions. The potency was expressed as an IC50 value of 38.379 μM. In comparison, acarbose had IC50 values of 34.216 μM.
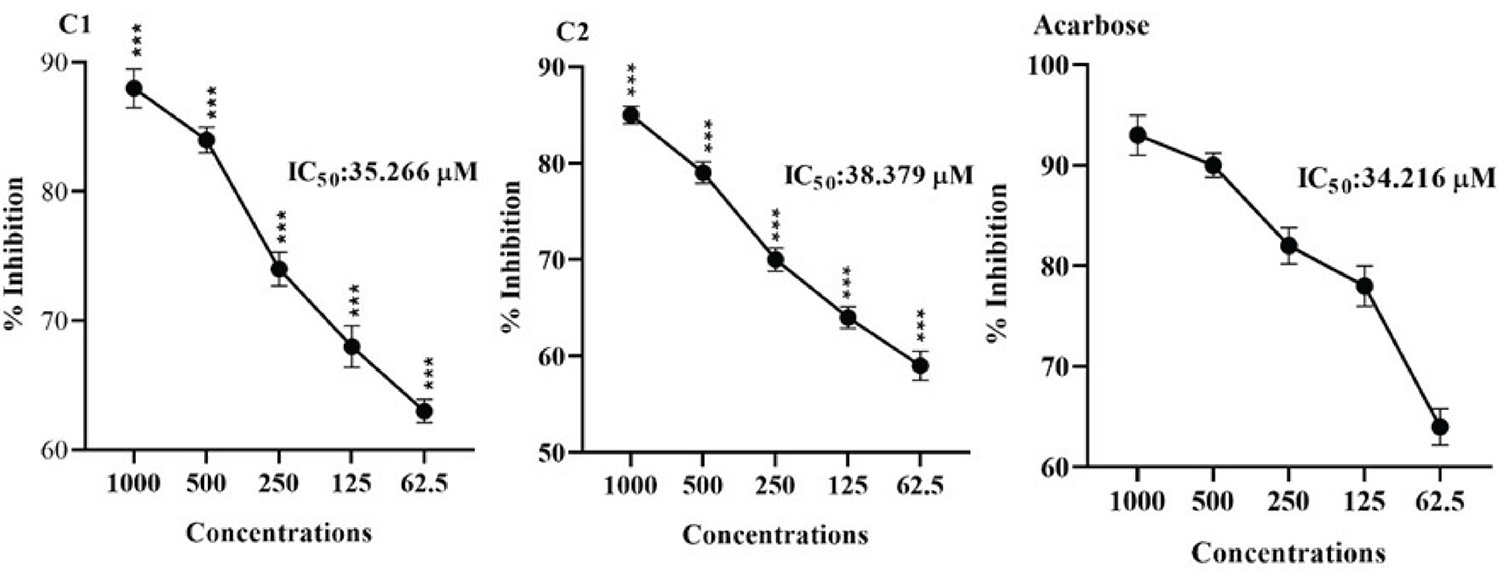
FIGURE 2 - Showing α-glucosidase inhibiting effects of Compounds (C1 and C2). The mean ± SEM of three distinct measurements was provided as the result. ANOVA at *p < 0.05 and the Bonferroni test showed static significance. At the same tested strengths, there are slight variations in ***p < 0.001 results when compared to the positive control (Acarbose).
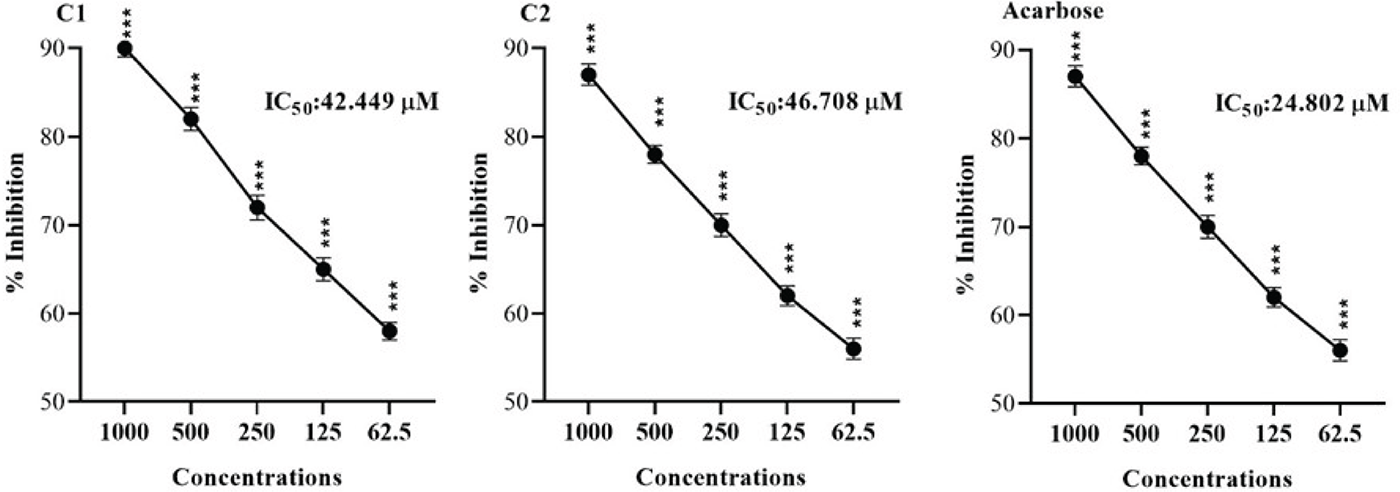
FIGURE 3 - Showing α-Amylase inhibiting effects of Compounds (C1 and C2). The Mean ± SEM of three distinct measurements was provided as the result. ANOVA at *p < 0.05 and the Bonferroni test showed static significance. At the same tested strengths, there are slight variations in ***p < 0.001 results when compared to the positive control (Acarbose).
Effect of Compounds on α-amylase Inhibition
The α-amylase inhibitory potential of test compounds (C1-C2) when challenged at various concentrations are displayed in Figure 3. Compound 1 caused significant inhibition of α-amylase, and the overall effects were concentration dependant. The maximum inhibition of 90% was observed at 1000 μM, and the IC50 was 42.449 μM. Similarly, compound 2 caused potential α-amylase inhibition at various doses from 62.50-1000 μM. The maximum inhibition, 87%, was demonstrated at 1000 μM. The overall effect of IC50 was 46.708 μM. The standard drug (Acarbose) had an IC50 value of 24.802 μM.
Molecular Docking Analysis
The binding free energies for ΔE are listed in (Table 1). The initial inhibitors have been adequately installed into their binding sites in order to attain their crystal configurations.
| No. | ΔG | RMSD | H. B | EInt. | Eele | LE |
|---|---|---|---|---|---|---|
| α-glucosides | ||||||
| C1 | −5.898 | 0.893 | −10.228 | −6.188 | −13.702 | −6.604 |
| C2 | −6.312 | 1.285 | −8.658 | −4.223 | −21.775 | −4.912 |
| Acarbose | −6.088 | 0.725 | −15.129 | −12.148 | −31.340 | −8.393 |
| α-amylase | ||||||
| C1 | −5.449 | 0.955 | 24.364 | −19.256 | −8.526 | 2.10 |
| C2 | −5.497 | 1.257 | 19.021 | −18.133 | −8.679 | 2.253 |
| Acarbose | −9.814 | 1.634 | -34.746 | −15.117 | −55.322 | 2.885 |
Where ΔG: Free binding energy of the ligand; RMSD: root-mean-square deviation; H.B.: H-bonding energy between protein and ligand; EInt. Binding affinity of H-bond interaction with receptor; Eele: Electrostatic interaction over the receptor, Ki: inhibition constant.
For the docking analysis, we selected the most perforable docking conformations of C1 and C2 compounds that were detected inside the active site of α-glucosidase enzyme (PDB: 3A4A) with proper alignment. The active site includes the hydrophilic amino acids (ASP69, TYR72, GLU277, HIS351, ASP352, and ARG442), which interact with competitive inhibitor acarbose. We noticed that almost all investigated compounds interacted with significant residues in the binding pocket. We defined the inhibitory behavior in terms of binding energy BE that was evaluated with the receptor. Then, redocked compounds compared the results to the comparative reference inhibitor and obtained a root mean square deviation (RMSD), which is summarized in (Table 1). The C2 exhibited strong interactions with the enzyme, as indicated by their binding free energies of −6.312 Kcal/mol. Compared to the reference inhibitor, which had a binding free energy of −6.088 Kcal/mol and lower inhibitory activity, the C1 showed superior binding efficacies, −5.898 Kcal/mol. The docking analysis revealed that C2 had the highest binding affinity and formed a strong hydrogen bond with Gly353, Glu411, and ASP307, which are essential for enzyme activity. The second inhibitor, C1, had a similar binding mode to the reference inhibitor and established a strong hydrogen bond with GLn353, ARG213, and Gln182 (Figure 4).
The molecule docked prolifically into active sites in a mechanism similar to that of the original inhibitors. For amylase, compounds C1 and C2 demonstrated promising binding affinities (ΔE = −5.44 to −5.5 kcal/mol), which substantiated their promising potencies. The interactions between the thirteen active compounds C1 and C2 with the active site residues were mainly polar bonds, hydrogen bonding, π−π, and π−H interactions, which contributed to a strong alignment with the enzyme backbone Figure 5. The active molecules C1 and C2 were attached deeply and comfortingly in the binding pocket by interacting with the hydrophilic Asp 198 hydrophobic binding pocket ALA197 and Ile 235. Compound C2 docked in the α-amylase binding pocket via hydrophobic interaction with Ala198. It is inferred that the formation of strong interactions with important residues can pinpoint α-amylase in the binding pocket. Lastly, according to the 3D-molecular docking, the C2 prefers a parallel orientation between the central indoline ring and the important hydrophilic Asp 197 into the binding pockets.
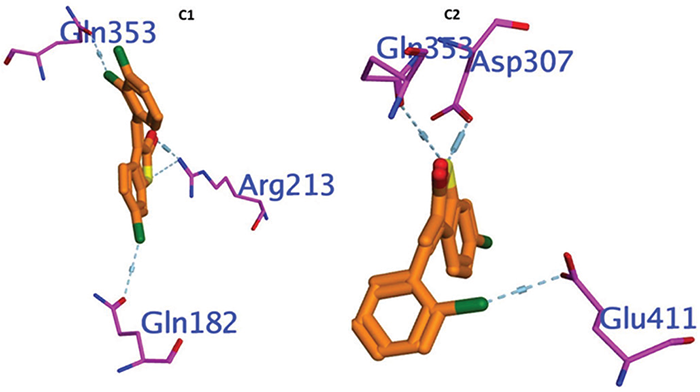
FIGURE 4 - Showing 3D Docking poses of C1, and C2 against α- glucosidase.
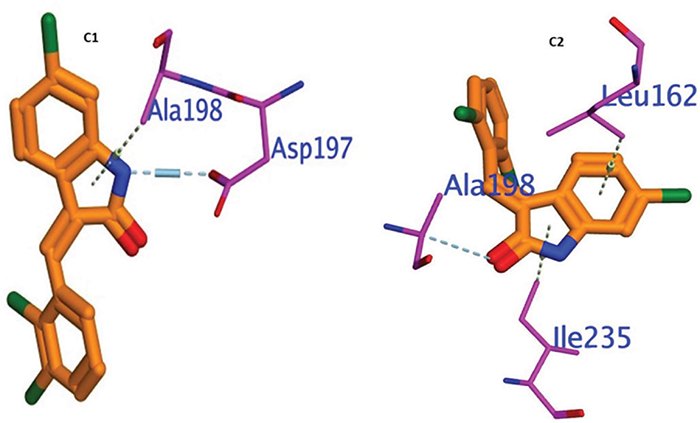
FIGURE 5 - Showing 3D Docking poses of C1, and C2 against α- amylase.
Molecular Dynamic Investigations of C1-C2
In order to comprehend enzymatic activity and create inhibitors, it is essential to investigate ligand-enzyme interactions, like the one that exists between the α-glucosidase protein and a ligand. Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) computations offer a thorough understanding of the molecular binding processes. The dynamic nature of molecular interactions is captured through a rigorous approach of accounting for the flexibility of α-glucosidase protein complexes during contact tests at 2 ns intervals. The energy changes in Figure 6, which illustrate fluctuations in the binding free energy at 12-ns intervals, provide important information about the strength and stability of the binding throughout time. The significance of structural variations on binding affinity is highlighted by comparing the energy changes of the two dichloroindolinone molecules. Positive interactions were indicated by the binding parameter values that are negative, which is encouraging for the possible effectiveness of these compounds as inhibitors. Effective binding is frequently indicated by a strong and stable hydrogen-bonding pattern, which is suggested by the minimal fluctuation in MM-PBSA values.
It is unsuitable for illustrating the relationship between molecular docking computations and ligand-protein interactions at the ns level since the ligand molecules are rather flexible in the calculations. The ligand-amylase protein interaction is performed at 250 ns using MM-PSBA calculations. In MM-PSBA calculations, all compounds of amylase protein complexes have flexibility when checking the interaction every 2 ns, and the energy change is reported in Figure 7. The resulting graph in Figure 7 depicts the binding free energy changes and variations in all ten (ns) intervals. The energy changes of the most active two 2, 3 and 2, 6-dichloroIndolinone molecules were compared, and the calculations were performed to validate the determination of bonding free energies using the molecular MMPBSA approach. Negative values for the relevant parameters indicate improved binding. The small fluctuations for MM-PBSA for both complexed with amylase over the entire time scale indicated a robust contact between Ligand and amino acid backbone, indicating a firm H-bonding pattern.
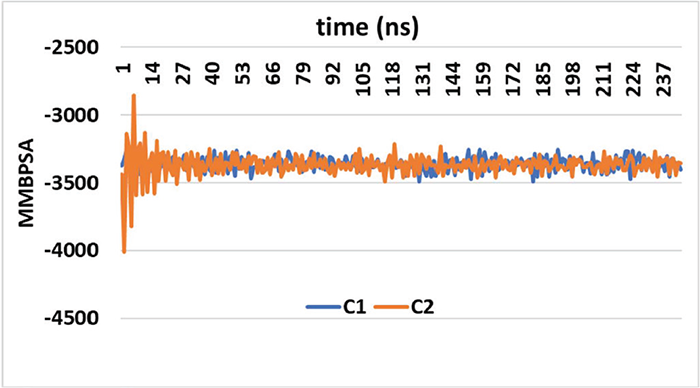
FIGURE 6 - The values of the test compounds C1 and C2 for the Gibbs free energy of ligand α-glucosidase protein.
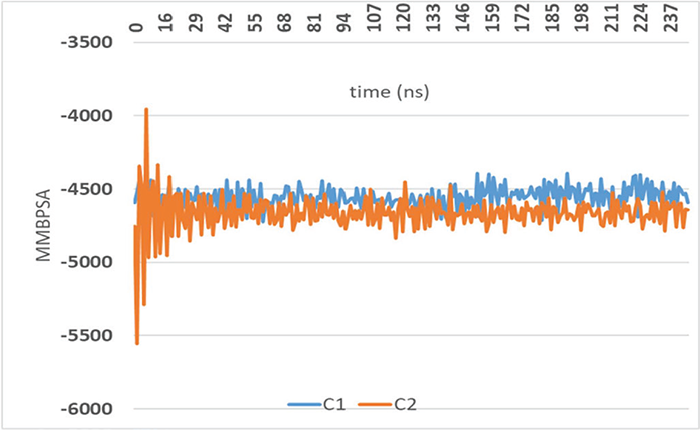
FIGURE 7 - The values of the test compounds C1 and C2 for the Gibbs free energy of ligand α-amylase protein.
Discussion
The study revealed significant in vitro α-glucosidase and α-amylase inhibition of oxindoles, 2, 3 dichloroIndolinone (C1) and 2, 6-dichloroIndolinone (C2), which were molecular docking and simulation studies.
The development of a new, more effective molecule with lower toxicity is always a primary target in research (21). Dichloroindolinone are oxindole derivatives that are utilized for a wide range of biological effects (10,22,23). Enzymes belonging to the glucosidases are present on the brush boundary surface of intestinal microglia. They cause starch 1,4-glycosidic bonds to break, releasing –D-glucose, which is absorbed. It consequently raises post-meal hyperglycemia. Therefore, it is essential to use these enzyme inhibitors in the management of type II diabetes. A class of enzymes known as α-glucosidases hydrolyzes the 1, 4-glycosidic bond in starch to produce α-D-glucose, which enters the bloodstream after absorption (24). As a result, it causes post-meal hyperglycemia. As a result, using these enzyme inhibitors is essential for managing T2DM. Proper absorption and digestion of monosaccharides may help avoid diabetes, obesity, hyperlipoproteinemia, and hyperlipidemia (25). These inhibitors may be used to treat malignancies, viral infections, and several other diseases, as well as to regulate the immune system (13). The early metabolism abnormality that arises in NIDDM, postprandial hyperglycemia, has been identified as a potential therapy for glucosidase (26). Our finding on the test compounds showed significant inhibition of the enzyme by both compounds and, therefore, could be potential candidates as glucosidase inhibitors.
The hydrolase enzyme α-amylase catalyzes the hydrolysis of internal -1, 4-glycosidic bonds in starch to create products like glucose and maltose. Since it is a calcium metalloenzyme, a metal cofactor is necessary for it to function (27). The class of enzymes known as amylases is responsible for hydrolyzing starch to produce low-molecular-weight dextrins and sugars, and they are crucial for the digestion of carbohydrates. Maltose, maltotriose, and other - (1,6) and - (1,4) oligoglucans can be broken down into glucose by various α-glucosidases and are the primary components of the final hydrolysate. Controlling the activity of HPA, which is a human pancreatic enzyme that is essential for the digestion of starch, could effectively lower blood glucose levels after meals (28). When combined, α-glucosidase and α-amylase inhibition can significantly reduce the rise in blood glucose that occurs after meals and could be an essential strategy for controlling blood glucose levels in people with T2DM. The main treatment targets for type-2 DM are thus pancreatic α-amylase and gut glucoamylases. Amylase antagonists are already available on the market for the treatment of diabetes. Amylase inhibitors can also be used to treat obesity (29). We observed strong inhibition of the α-amylase by the test compounds at various concentrations and therefore augmented the overall anti-diabetic potential of these compounds as strong candidates for further studies.
Molecular docking tests revealed that the best-ranked conformation of the test compounds fit into the site of activity well, and its oxindole moiety interacted with important locations in the binding pocket. The interaction between ligands and enzymes was studied in docking investigations to gain a greater understanding of the in vitro outcomes. The structure data for the selected enzymes was obtained by searching the Protein Data Bank. The PDB IDs for α- Amylase and α-Glucosidase were 4GQR and 5NN5, respectively (28,30). The process and enzyme preparation were completed with the aid of the Molecular Operating Environment (MOE) software. The enzymes were first removed from non-standard structures and inadequate bounded ligands in order to prepare them for docking in PDB format. Binding energies were investigated, and docking method outcomes were evaluated. It was demonstrated that the optimal ligand-enzyme interaction model produced increased negative values; the more negative values, the greater the probability of binding at that particular site. Various situations were used in order to see the key interaction between the ligands and enzymes, amino acid residues, binding pocket, and bond lengths were presented in different colors. The lowest score poses, and RMSD revealed increased stability in the binding pocket. The data was utilized to rank the docked poses and to select the most capable docked conformation of each compound. The hetero ring for Erlotinib and benzimidazole centered on the α-amylase pocket and interacted with HIS 305, ASP 300, AlA198, ASP 197, THR 163, LEU162, TYR 151, GLN 63, TRP 59. In the MM-PBSA calculations, the ligand-glucosidase and amylase protein interaction is monitored every 2 ns. The negative values indicate improved binding (more favorable interactions). The small fluctuations observed for MM-PBSA over the entire time scale suggest a stable interaction. The robust contact between the ligand and the amino acid backbone indicates firm hydrogen bonding.
Conclusion
In a dose-dependent way, the 6-chlorooxindole-derived compounds C1 and C2 showed potential in vitro α-glucosidase and α-amylase inhibition. C1 and C2 demonstrated a high binding and inhibitory potential when docked with the enzyme α-Glucosidase (5NN5) and α-amylase (4GQR) enzyme. Strong alignment with the enzyme backbone was achieved by the active chemicals C1 and C2 interacting primarily with the active site residues through polar bonds, hydrogen bonding, π−π, and π−H interactions. The minimal fluctuation in MM-PBSA values suggests a consistent and strong hydrogen-bonding pattern, which is often a hallmark of effective binding. The present investigation reveals improved anti-diabetic features for the 6-chlorooxindole derivatives. These focused compounds could help develop more potent antihyperglycemic medications in the future, but need further detailed studies.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2025R812), King Saud University, Riyadh, Saudi Arabia, for funding.
Disclosures
Financial support: The authors extend their appreciation to the Researchers Supporting Project number (RSPD2025R812), King Saud University, Riyadh, Saudi Arabia, for funding.
Conflict of Interest: The authors of this article declare no conflict of interest.
Data Availability Statement: The data that support the findings of the present study are available from the corresponding authors upon reasonable request.
References
- 1. Sagbo IJ, van de Venter M, Koekemoer T, et al. In vitro anti-diabetic activity and mechanism of action of Brachylaena elliptica (Thunb.) DC. Evidence-Based Complementary and Alternative Medicine, 2018. CrossRef PubMed
- 2. Galicia-Garcia U, Benito-Vicente A, Jebari S, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275. CrossRef PubMed
- 3. Khan MAB, Hashim MJ, King JK, et al. Epidemiology of type 2 diabetes—global burden of disease and forecasted trends. J Epidemiol Glob Health. 2020;10(1):107-111. CrossRef PubMed
- 4. Pozzilli P, Di Mario U. Autoimmune diabetes not requiring insulin at diagnosis (latent autoimmune diabetes of the adult): definition, characterization, and potential prevention. Diabetes Care. 2001;24(8):1460-1467. CrossRef PubMed
- 5. Resnick HE, Howard BV. Diabetes and cardiovascular disease. Annu Rev Med. 2002;53(1):245-267. CrossRef PubMed
- 6. Khursheed R, Singh SK, Wadhwa S, et al. Treatment strategies against diabetes: success so far and challenges ahead. Eur J Pharmacol. 2019;862:172625. CrossRef PubMed
- 7. Khetmalis YM, Shivani M, Murugesan S, et al. Oxindole and its derivatives: a review on recent progress in biological activities. Biomed Pharmacother. 2021;141:111842. CrossRef PubMed
- 8. Gassman PG, Van Bergen T. General method for the synthesis of oxindoles. J Am Chem Soc. 1973;95(8):2718-2719. CrossRef
- 9. Mukhliss L, Taha M, Rahim F, et al. Synthesis and novel structural hybrid analogs of oxindole derivatives bearing piperidine ring, their antidiabetics II activity and molecular docking study. J Mol Struct. 2025;1332:141666. CrossRef
- 10. Pirzada AS, Khan H, Alam W, et al. Physicochemical properties, pharmacokinetic studies, DFT approach, and antioxidant activity of nitro and chloro indolinone derivatives. Front Chem. 2024;12:1360719. CrossRef PubMed
- 11. Ahmad I, Khan H, Serdaroğlu G. Physicochemical properties, drug likeness, ADMET, DFT studies, and in vitro antioxidant activity of oxindole derivatives. Comput Biol Chem. 2023;104:107861. CrossRef PubMed
- 12. Kadhim MM, Heravi MRP, Mohammadi-Aghdam S, et al. Investigation of anti-tumor (E)-3-X-oxindole via functionalization of C20 nano structure: A DFT approach. Comput Theor Chem. 2022;1214:113763. CrossRef
- 13. Khan M, Yousaf M, Wadood A, et al. Discovery of novel oxindole derivatives as potent α-glucosidase inhibitors. Bioorg Med Chem. 2014;22(13):3441-3448. CrossRef PubMed
- 14. Altowyan MS, Barakat A, Al-Majid AM, et al. Spiroindolone analogues as potential hypoglycemic with dual inhibitory activity on α-amylase and α-glucosidase. Molecules. 2019;24(12):2342. CrossRef PubMed
- 15. Rauf A, Khan H, Khan M, et al. In Silico, Swiss ADME, and DFT studies of newly synthesized oxindole derivatives followed by antioxidant studies. J Chem. 2023;2023(1):5553913. CrossRef
- 16. Huneif MA, Alshehri DB, Alshaibari KS, et al. Design, synthesis and bioevaluation of new vanillin hybrid as multitarget inhibitor of α-glucosidase, α-amylase, PTP-1B and DPP4 for the treatment of type-II diabetes. Biomed Pharmacother. 2022;150:113038. CrossRef PubMed
- 17. Taha M, Ismail NH, Khan A, et al. Synthesis of novel derivatives of oxindole, their urease inhibition and molecular docking studies. Bioorg Med Chem Lett. 2015;25(16):3285-3289. CrossRef PubMed
- 18. Irfan A, Faisal S, Ahmad S, et al. Structure-based virtual screening of furan-1, 3, 4-oxadiazole tethered N-phenylacetamide derivatives as novel class of hTYR and hTYRP1 inhibitors. pharmaceuticals, 2023, 16, (3), 344. CrossRef
- 19. Nahoum V, Roux G, Anton V, et al. Crystal structures of human pancreatic α-amylase in complex with carbohydrate and proteinaceous inhibitors. Biochem J. 2000;346(Pt 1):201-208. CrossRef PubMed
- 20. Yamamoto K, Miyake H, Kusunoki M, et al. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010;277(20):4205-4214. CrossRef PubMed
- 21. Schenone M, Dančík V, Wagner BK, et al. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9(4):232-240. CrossRef PubMed
- 22. Rindhe SS, Karale BK, Gupta RC, et al. Synthesis, antimicrobial and antioxidant activity of some oxindoles. Indian J Pharm Sci. 2011;73(3):292-296. PubMed
- 23. Qi Y, Gong H, Wang Z, et al. Discovery of novel oxindole derivatives as TRPA1 antagonists with potent analgesic activity for pain treatment. Bioorganic Chemistry 154, 108088.
- 24. Casirola DM, Ferraris RP. α-Glucosidase inhibitors prevent diet-induced increases in intestinal sugar transport in diabetic mice. Metabolism. 2006;55(6):832-841. CrossRef PubMed
- 25. Derosa G, Maffioli P. α-Glucosidase inhibitors and their use in clinical practice. Arch Med Sci. 2012;8(5):899-906. CrossRef PubMed
- 26. Thilagam E, Parimaladevi B, Kumarappan C, et al. α-Glucosidase and α-amylase inhibitory activity of Senna surattensis. J Acupunct Meridian Stud. 2013;6(1):24-30. CrossRef PubMed
- 27. de Souza PM, de Oliveira Magalhães P. Application of microbial α-amylase in industry - a review. Braz J Microbiol. 2010;41(4), 850-861. CrossRef PubMed
- 28. Qin X, Ren L, Yang X, et al. Structures of human pancreatic α-amylase in complex with acarviostatins: implications for drug design against type II diabetes. J Struct Biol. 2011;174(1):196-202. CrossRef PubMed
- 29. Jayaraj S, Suresh S, Kadeppagari RK. Amylase inhibitors and their biomedical applications. Stärke. 2013;65(7‐8):535-542. CrossRef
- 30. Azam SS, Uddin R, Wadood A. Structure and dynamics of alpha-glucosidase through molecular dynamics simulation studies. J Mol Liq. 2012;174:58-62. CrossRef