|
AboutOpen | 2019; 5(1); 70-75
POINT OF VIEW |
Getting your devices ready for MDR compliance – a clinical approach and orthopaedic device manufacturers’ perspective
Abstract
On 26 May 2017, the Medical Devices Regulation (MDR 2017/745) was published, with the aim of replacing the current Medical Devices Directive (MDD 93/43/EC). Following a transition period of 3 years, this regulation will be applied in full from 26 May 2020. This article describes and discusses the impact of MDR on the clinical evaluation and gives advice to manufacturers on the preparation, planning and implementation of processes to support compliance with the new regulation.
Keywords: Clinical evaluation, Medical Devices Regulation, Orthopaedics, Post-market clinical follow-up, Variant
Submitted: May 31, 2019
Accepted: July 11, 2019
Published online: July 31, 2019
© 2019 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0). Any commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Introduction
There have been some significant alterations to the European Medical Devices Directive (MDD) 93/42/EEC and the Active Implantable Medical Device (AIMD) Directive 90/385/EEC. These will result in the harmonisation of clinical investigation regimens and strict standards for the certification of both existing historic and new medical devices. The changes will also ensure that compliance of medical devices continues post-marketing. Medical Devices Regulation (MDR, MDR 2017/745) replaced the current MDD on 26 May 2017. Following a transition period of 3 years, MDR regulation will be applied fully from 26 May 2020, which is known as the Date of Application.
This article details our analysis of how the clinical evaluation work stream will be affected by the new MDR. We will also offer advice to manufacturers regarding the preparation, planning and implementation of MDR-compliant processes during the transition period. As specialists from the orthopaedics industry, our views do not necessarily reflect the views of other stakeholders such as the Notified Bodies.
Clinical evaluation is necessary to verify the safety and performance of a medical device. This involves a structured, continual process of collecting and evaluating relevant clinical data. In recent years, the clinical evaluation of medical devices has become more refined and more central to regulatory processes. The European guidance on clinical evaluation, MEDDEV 2.7/1 revision 3, was replaced by revision 4 in 2016. This introduced some significant changes that had an impact on manufacturers immediately. The current MDD gives the Clinical Evaluation Report (CER) a central role with the conformity assessment of a device, and it is a requirement for the Conformité Européenne (CE) marking. The CER includes appraisal and analysis of clinical data to support conclusions about the safety and performance of devices. Regular and proactive updates are required, but many manufacturers mistakenly believe that merely “passing” the initial Notified Body review is sufficient and that limited maintenance is necessary. The MDR challenges this assumption, and a CER that has withstood scrutiny once will not necessarily pass subsequent assessments because there might be recommendations for improvement to include new and additional data from post-marketing phase. This is applicable to both high-risk and low-risk devices.
The MDR’s requirement for more detailed assessments means that manufacturers must urgently assess their practices of maintaining and updating CERs. In this article, we will describe how the substantiation of medical device usage can be supported by proper planning that incorporates a gap analysis, proactive collection of clinical data and appropriate published research.
Performing the gap analysis
The new unified European regimen for clinical evaluations and investigations under the MDR contrasts with the current system in which member states have different regulations. There will also be mandatory post-market clinical follow-up (PMCF) and periodic safety update reports (PSUR). The MDR requires manufacturers’ clinical evaluation strategy, post-market surveillance (PMS) and PMCF to be rigorously reviewed by the Notified Body. Manufacturers will also have to conduct PMCFs (unless they can justify why they are not necessary) and provide post-market clinical data and information about the safety, performance and clinical benefits of devices according to the associated level of risk. Therefore, gap analyses will be required to identify any gaps in the clinical evidence for devices on the market, followed by appropriate updates to clinical strategy and data. Here are some of the strategies that will ensure compliance with the MDR.
Proper planning
There will be no carrying over from MDD (grandfathering), so all CE-marked devices on the market will have to be assessed according to the new MDR requirements. Most medical devices will remain in the same classification, but a small minority will be subject to classification change. Spinal disc replacement implants, for example, will be in Class III. However, it should be noted that legacy devices that are not reclassified will not necessarily have a quick or easy route to compliance.
It is essential to understand the differences between the MDR and the current MDD. The General Safety and Performance Requirements (GSPRs), which is Annex I of the MDR, will identify new conditions that need to be addressed for most CE-marked legacy devices. Clinical data, labelling and technical documentation might also need to be updated. The new requirements could also necessitate careful reviews and updates to quality assurance, risk management, PMS and other regulatory and quality processes.
Conducting a full review
General considerations
Once the MDR has been fully implemented, medical device manufacturers will have to meet the GSPRs of Annex I. Grandfathering of legacy (pre-MDR) devices will not be allowed, so all CE-marked medical devices will have to be fully MDR compliant.
The MDR is very detailed, and each medical device will be subjected to review of the Technical Documentation, including instructions for use (IFU) to determine whether its current use needs to be updated or abandoned. To ensure that manufacturers are compliant and that safety, performance and clinical benefits of the devices are fully supported by sufficient clinical and technical data and documentation, Notified Bodies will conduct comprehensive and detailed reviews. Because the MDR will have more stringent requirements for acceptable clinical evidence, it might be necessary to acquire new and/or additional data. With legacy devices undergoing updated, robust clinical evaluations, there might be some devices that are deemed not commercially profitable enough to justify the resources required to make them MDR compliant.
Comprehensive gap analyses will reveal how the new MDR requirements will affect both legacy and new devices, and this will guide your efforts and planning.
The requirements of clinical evaluation, investigation and reporting have all been defined by the MDR. Scientific principles will have to underpin any evaluation of clinical data according to a pre-defined procedure. A critical assessment of the relevant scientific literature, in relation to the safety, performance, clinical benefits, design characteristics and purpose of the device, will have to form the basis of clinical evaluation. Alternative treatment options will also have to be taken into account. Manufacturers will have to demonstrate that the data supports the intended purpose of the device and compliance with GSPRs.
The CER and current clinical data must also be assessed to ensure MDR compliance. Notified Bodies are responsible for ensuring a higher level of quality of clinical evidence, so manufacturers will be expected to deliver compliance with clinical data requirements.
The MDR will mean more intense scrutiny of device equivalence comparisons for both low-risk and high-risk (Class III and implantable) devices. Manufacturers will be required to demonstrate that the new device (subject device) has the same characteristics – technically, biologically and clinically – as the intended equivalent device, and that there is sufficient access to the equivalent device’s technical information. For the high-risk devices (Class III and implantable), they will have to provide an agreement to access the equivalent device’s technical documentation if it belongs to a different manufacturer.
Manufacturers should plan for the challenges in acquiring the necessary data for the CER of their medical devices well in advance. Assessing the key products is a good first practical step. A manufacturer’s portfolio can be assessed according to the following matrix (Tab. I):
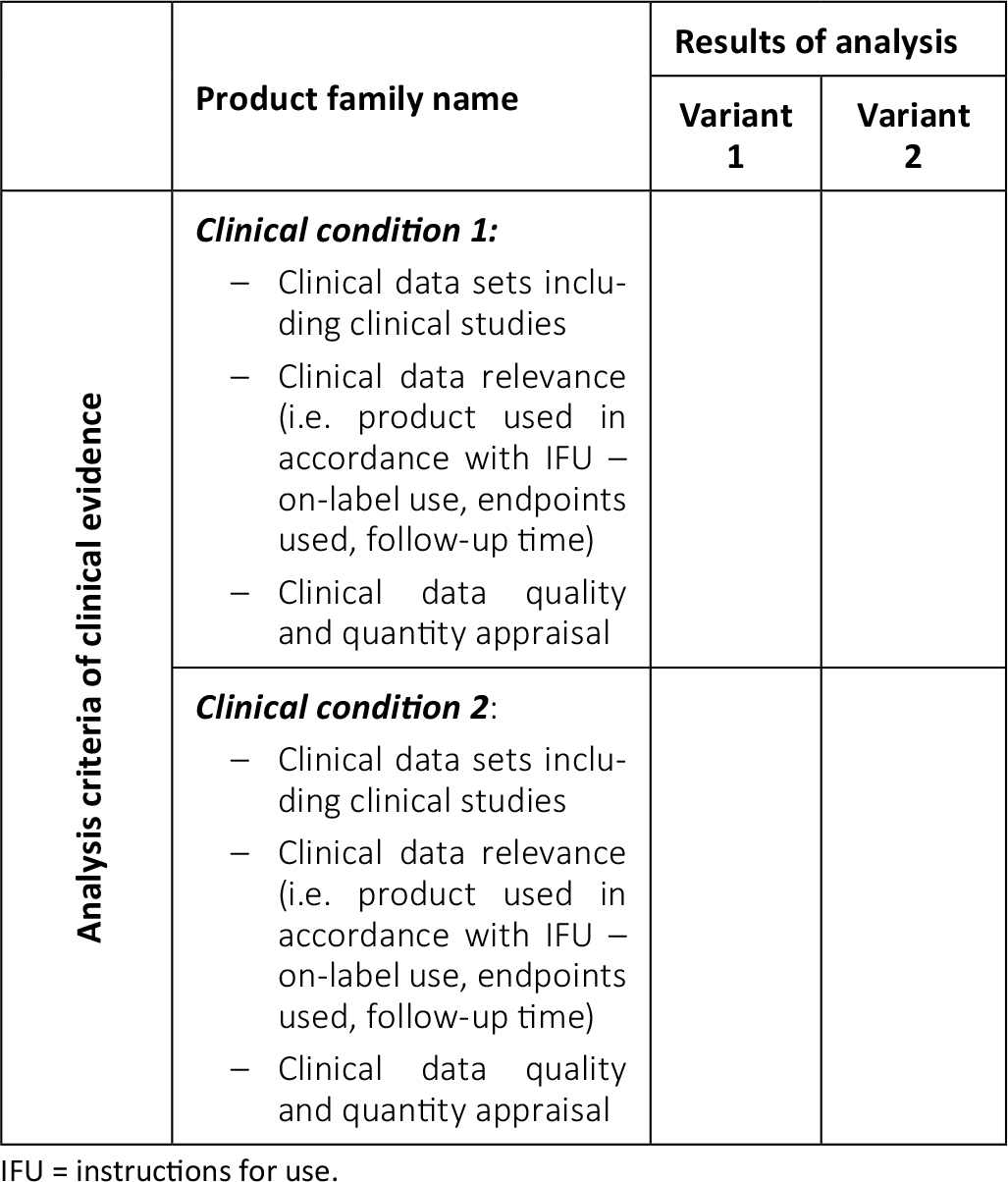
Definition of variant
Our definition of a “variant” is a subset of a product family where different clinical outcomes cannot be ruled out. For example, in total hip arthroplasty, a hip stem family could have both uncemented and cemented variants. Uncemented stems are made of porous alloys like titanium, while most cemented stems are made of either cobalt chrome or stainless steel alloys. These two stem variants differ substantially in terms of deployment methods: the first variant is used in the absence of bone cement as a stand-alone, while the second variant is used with bone cement to promote fixation. Different sizes and/or offsets of a hip stem are not usually considered to be product variants though. These different sizes and/or offsets offer the orthopaedic surgeon flexibility with reconstructing different patients’ anatomies. If the anatomy is well reconstructed, clinical outcomes should be the same, regardless of stem sizes and/or offsets. Within risk management, analysis of the impact of different sizes and/or offsets is made. Once it has been established that there are no different residual risks associated with these sizes and/or offsets, we can treat them as a single variant within the clinical evaluation.
Clinical conditions – indications
Clinical indications should be clearly described in the IFU of the medical device. For example, in total hip arthroplasty, a device (hip stem or acetabular component) might perform well in a patient with relatively healthy bone, adequate bone stock, etc., but the design of the same implant may not be optimal for adequate fixation when implanted in a revision setting where there is poor bone quality and insufficient bone stock. Similar clinical outcomes cannot therefore be assumed. In this case, there are two different clinical conditions and two different indications (“primary” and “real” revision), so both of them will require clinical data. If the device is indicated for use in similar initial bony conditions (primary clinical condition with good bone stock and quality), a similar clinical behaviour can be expected. Therefore, we can assume that the clinical data of one indication (primary) applies to the other (i.e. early revision that is “primary-like” clinical condition).
Another example in the field of hip arthroplasty is the use of acetabular liners for primary implantations and hip revision arthroplasty in cases where the acetabular component is well fixed. In most circumstances, revising a well-fixed acetabular component is obviously neither beneficial to the patient nor cost-effective. The orthopaedic community considers an isolated revision of the acetabular liner to be state-of-the-art. Therefore, we recommend that the two usages are not considered to be two different clinical conditions, and we would only use data from the primary condition to substantiate the revision indication’s safety, performance and clinical benefits. We should also question whether it is ethical to perform research studies where the answer is already known. In the case of an acetabular liner, for example, a similar performance (minimal polyethylene wear) can be expected in both primary and revision procedures (assuming that the acetabular component is well fixed and only the liner needs to be exchanged). Therefore, within the framework of a clinical study, it is highly questionable whether clinical performance data (polyethylene wear and its consequences) for the revision indication of acetabular liners should be collected at all when it is already available in the primary indication. Assertions like this should always be aligned with the state-of-the-art and subjected to risk management. Manufacturers argue to keep this indication in the IFU as the alternative to revising a well-fixed acetabular component, which could lead to additional risk and harm to the patient. Clinical studies should be designed to provide scientifically and ethically relevant data (1, 2), and it may not be in the interests of patients to be part of studies that do not generate relevant clinical data. Alternatives should therefore be carefully considered by all stakeholders, including regulators, manufacturers and healthcare professionals.
Hemiarthroplasty is another example that illustrates this point. It is an established and accepted surgical procedure that involves the implantation of only a hip stem and ball head. Because the procedure is typically performed in elderly, fragile and vulnerable patients with limited life expectancy, it is difficult to follow-up over a longer period of time, and it is therefore legitimate to question whether a clinical study will be a burden on them. In these cases, inferences on the performance of the devices (hip stem and ball head) could be more appropriately generalised from other indications (primary total hip arthroplasty). We therefore recommend that regulators prioritise patients’ safety and benefits by proactively seeking advice from clinicians and healthcare professionals and tailoring some of the regulatory requirements to the specific clinical conditions.
MDR Article 61 point 10 supports this interpretation. It states that the demonstration of conformity with GSPR not based on clinical data may be appropriate if it is based on the findings of the manufacturer’s risk management.
Under the MDD, clinical evidence was required, with low-risk devices often supported by clinical data of equivalent devices, and high-risk devices requiring original clinical data. However, the MDR will require more stringent CERs (see EU-MDR Annex XIV, Part A). The public Summary of Safety and Clinical Performance will be required for implantable devices and Class III devices (excluding custom-made or investigational devices) (EU-MDR Article 32). In addition, clinical data will mostly need to derive from clinical investigations or PMCFs that have followed established good clinical practices, such as ISO 14155:2011 (2) and the World Medical Association Declaration of Helsinki (1), and been performed under the supervision of a sponsor.
The MDR allows for the possibility of submitting one central application for conducting clinical investigations performed in multiple European countries. This is a positive step, but will not be in effect until a later date.
Under the MDR, using device equivalence route in CERs will be more difficult, particularly for Class III and implantable devices. Therefore, it is worth assessing the value of pursuing equivalence in the Class III device CER, especially where there is no contractual arrangement for full access to competitors’ technical documentation. In these cases, we recommend planning a clinical investigation because it will generate clinical data that is directly applicable to your device.
Manufacturers are advised to review their entire product portfolio and associated CERs in order to ensure that appropriate clinical strategies have been adopted in line with EU-MDR compliance. As previously stated, devices will probably require proactive data collection (PMS/PMCF) where there is a lack of clinical evidence.
Some devices may require amendments and revisions to their IFU, and manufacturers may have to abandon certain indications for use where there is little or no supporting clinical evidence.
Time management and resource allocation
Manufacturers must allow sufficient time for the planning stage. Gap assessments will establish what work will be needed and whether PMCF/PMS studies are warranted or current equivalence is sufficient. Additional work will be required for both equivalence and PMCF/PMS, so resource allocation and time management are critical. Planning work should have begun already.
Gap assessments of pre-MDR legacy devices will be similar to premarket evaluations. They should include general safety and performance data, technical data, clinical evidence (including any planned and ongoing PMCF), risk assessments, Unique Device Identification (UDI), labelling and technical documentation, among other elements. Post-market activities will also be affected in areas such as vigilance and surveillance requirements.
Once a gap assessment has been completed, the implementation phase, in which the actual work to address the gaps, should commence. Additional data and documentation will probably be needed to fulfil new MDR requirements, so changes to systems and procedures will be necessary. This change management takes time and might involve the hiring of staff and/or technical vendors in different functional areas, so proper time management is essential. Financial planning will also be necessary.
Relevant stakeholders, including device manufacturers, Notified Bodies and Competent Authorities, will be on a collective learning curve as the new MDR requirements come into force. Potential issues with compiling, reviewing and accepting data, as well as calibrating to the new level of data, will need to be anticipated. Accommodating these eventualities will require additional time.
We would urge manufacturers to take immediate action. It might be tempting to wait and analyse the process after the initial reviews have been made, but this course of action would be unwise in the case of the transition to EU-MDR. We strongly recommend that manufacturers communicate, analyse, make decisions and act quickly and efficiently to become EU-MDR compliant at the earliest opportunity.
If you have not already done so, communicating with the relevant Notified Body is the first step. Your company’s interpretation of the requirements may differ from that of the Notified Body. If you communicate openly and continuously, goodwill will prevail, and you will receive valuable feedback. The Notified Bodies will probably be overloaded because of the predicted shortage, so you should start this dialogue immediately. You might also be able to get valuable information from medical industry associations, online resources, webinars and other white papers provided by the Notified Bodies.
The state-of-the-art section of CER
It is essential to have a sound description of the state-of-the-art, as this sets a benchmark for the device’s clinical performance. In EN ISO 14971: 2012, D.4 state-of-the-art is defined as “what is currently and generally accepted as good practice” (3). There are various ways to determine the state-of-the-art for a medical device, including standards and best practice applied to the products that are the same or similar, and the findings of recognised scientific research.
To determine the state-of-the-art based on this definition, it is necessary to apply a combination of clinical practice guidelines and recommendations (cf. norms), register evaluations (cf. best practice) and results of literature research (cf. scientific research). Other useful references are Competent Authority websites and registries, health technology assessment reports, systematic review databases (e.g. Cochrane) and practice and consensus guidelines. Describing the state-of-the-art in the CER has become much more important with the introduction of MEDDEV 2.1/7 rev. 4. A literature search must be carried out to determine the state-of-the-art for a device, and this search should be separate from the literature search that was performed to evaluate the device because the scope and methodology differ. This search needs to be broader and not limited to evaluating the subject device.
Your CER’s state-of-the-art section should generally contain the following elements:
- The condition that is being treated
- Epidemiology data
- Methods for classifying and managing the condition
- Justifications for the choice of clinical endpoints
- Potential clinical hazards
- The “gold standard” treatment
- The alternative forms of treatment for the condition
- The alternative treatments within different patient groups
- Risk assessment of alternative treatments
- A comparison between the state-of-the-art and the subject device
In many cases, however, the state-of-the-art is either poorly described or not subject to any scientifically valid studies at all. Often, there is a lack of scientific evidence to assess the merits of various treatment strategies. Ideally, clinical decision-making is based on randomised clinical trials (RCTs) that are well-designed and well-conducted, and the generalisability of the RCT has been confirmed by replicating it under similar or different conditions. Although scientific purists will only accept scientific evidence for clinical decision-making, those on the front line frequently have to make judgements in the absence of perfect data. The British Medical Journal illustrated this point in a 2008 paper that argued the effectiveness of parachutes has not been subjected to rigorous evaluation by using RCTs (4). This article concluded that “common sense might be applied when considering the potential risks and benefits of interventions” in the absence of perfect scientific data. The authors argued that the only alternative is “to continue the quest for the Holy Grail of exclusively evidence-based interventions and preclude parachute use outside the context of a properly conducted trial” (4). Regulators, clinicians and manufacturers should be aware of this pitfall and consider whether a study makes sense and the relevancy of evidence-based proof of a state-of-the-art.
Therefore, it is essential to consider sources other than the scientific literature. In addition to scientific evidence, the authors of this article recommend including appraisals that are based on the expertise and clinical judgement from national registries, consensus groups, national organisations (such as the UK’s National Institute for Health and Care Excellence) and healthcare professional associations (such as the European Federation of National Associations of Orthopaedics and Traumatology).
Clinical data
According to the MDR, clinical data is defined as information concerning the safety and performance of a device that is generated from its use. When conducting clinical evaluations for a device, three main sources will be included:
- Unpublished internal manufacturer’s data (clinical investigation or PMCF)
- Studies that have been published in medical journals
- Complaints and post-market data
For certain types of devices, registry data should be included as a fourth data source for the CER. For example, in the case of orthopaedic implants, several national joint registries monitor outcome data, such as the annual rate of revisions. Registries collect data on large numbers of patients, so they offer an unrivalled statistical power and facilitate robust methodologic approaches that would not be possible with PMCF studies. Therefore, registries are a reliable and good-quality source of verifying any differences in outcomes related to a device’s design characteristics. For example, in arthroplasty, PMCF studies are not sufficient to measure relatively small differences in outcomes that could take place over a longer period of time. Registries are also better equipped than PMCF studies to assess confounders such as factors at the level of patient, surgeon or hospital (5, 6). In many cases, registries have been an indispensable source of information for manufacturers, healthcare institutions and policymakers.
Studies published in peer-reviewed medical journals are also good sources of data because they should be based on rigorous clinical trial methodology and processes that have been vetted by the publication’s reviewers and editors.
PMCF was once considered merely a “check-the-box” activity, but it is now being taken much more seriously. Annex XIV Part B of the EU-MDR is devoted to PMCF, and Notified Bodies now look for PMCF processes that are proactive and “living”.
Your clinical evaluation will be enhanced by data from previous, ongoing clinical investigations and PMCF studies. In order to bring your CER to the required level, this data must be continuously and critically re-evaluated. Compliance with EN ISO 14155:2011 is a necessity. Measures must be put in place to avoid any bias. This could include randomisation, consecutive series study design, monitoring/auditing, appropriate data collection methods and objective endpoints.
PMCF will be majorly impacted by the MDR’s new requirements for higher quality of clinical data. Marketing claims will have to be corroborated by data related to the device in question or an equivalent device. Under the MDR, there might be challenges to previous studies that do not comply with current ethical standards. This could affect many CE-marked devices because they might have referenced older equivalent devices that did not meet the MDR requirements. The availability and quality of existing clinical data should therefore be established as part of the planning process for the transition to the MDR.
Notably, during the grace period, products that are certified under MDD are not exempted from MDR’s increased clinical evidence requirement. Specifically, MDR requires PSUR (periodic safety update report), that is, summarising the results and conclusions of the analyses of the PMS data (including PMCF) gathered as a result of the PMS plan, and this requirement also applies to products certified under the MDD.
Data that was compliant with the MDD will not necessarily be compliant with the MDR. It is possible that some long-established devices that are compliant with the MDD will see their clinical evaluation invalidated because they do not reach the higher threshold for medical scientific relevance.
In many instances, the scientific literature will be scant, so it is likely that PMCF will be expected to be performed on well-established, generic products, such as wires, screws, pins, sutures and plates in the case of orthopaedics. The medical and academic community rarely conducts scientifically unoriginal studies for regulatory purposes, so this will be a challenging process. Most investigators will be unwilling to submit their studies on wires, bone screws, pins, sutures or plates to scientific journals, and most journals will be reluctant to publish such articles because they have no scientific relevance. These products may have been on the market for decades, and their performance is well known, so there might be little to gain in terms of scientific knowledge. Ethics Committees will be unlikely to approve these types of studies because there is no genuine scientific interest and no patient benefit. The most established products have the least amount of scientific evidence, so they might be unable to withstand the scrutiny of Notified Bodies and Competent Authorities. It is not yet clear how much the MDR solely relies on clinical data or whether it will be acceptable to demonstrate conformity with GSPR not based on clinical data. To support decision making, Common Specifications should be developed under the MDR for medical devices that are currently deemed well-established technology and considered to be state-of-the-art.
Conclusions
The European regulatory landscape faces significant change under the MDR, particularly with the increased focus on the clinical evaluation, clinical data and reporting. Manufacturers need to take immediate action, and this should include gap analyses and updates to procedures, systems and resources. The MDR will lead to an increase in the number of clinical studies, but, in the opinion of the authors of this article, the initiation of clinical studies that lack scientific relevance should be challenged. To illustrate this, we presented and discussed examples from the field of orthopaedics.
Disclosures
Financial support: None of the authors have any financial interest or benefit arising from this work and publication.
Conflict of interest: PF is a consultant for Zimmer Biomet, although no payments related to this work were received from Zimmer Biomet. HA is an employee of Zimmer Biomet.
References
- 1. World Medical Association. WMA Declaration of Helsinki. Ethical principles for medical research involving human subjects. 64th WMA General Assembly, Fortaleza, Brazil; 2013.
- 2. European Committee for Standardization. Clinical investigation of medical devices for human subjects – good clinical practice (ISO 14155:2011); 2011.
- 3. European Committee for Standardization. Medical devices – application of risk management to medical devices (ISO 14971:2007, Corrected version 2007-10-01). vol EN ISO 14971; 2012.
- 4. Smith GCS, Pell JP. Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ. 2003;327(7429):1459-1461.
- 5. Pugely AJ, Martin CT, Harwood J, Ong KL, Bozic KJ, Callaghan JJ. Database and registry research in orthopaedic surgery: part 2: clinical registry data. J Bone Joint Surg Am. 2015;97(21):1799-1808.
- 6. Labek G, Neumann D, Agreiter M, Schuh R, Bohler N. Impact of implant developers on published outcome and reproducibility of cohort-based clinical studies in arthroplasty. J Bone Joint Surg Am. 2011;93(Suppl 3):55-61.