|
AboutOpen | 2023; 10: 35-42 ISSN 2465-2628 | DOI: 10.33393/ao.2023.2513 BRIEF REPORT |
X-linked hypophosphatemic rickets: cases series and literature review with a focus on neurosurgical management
Abstract
X-linked hypophosphatemic rickets (XLH) is an X-linked dominant disease caused by mutations in the Phosphate-Regulating Endopeptidase X-Linked (PHEX) gene. Due to its rarity and the wide range of clinical manifestations, management of the disease can be challenging due to several associated clinical implications that may arise during follow-up. The neurological complications associated with XLH are the most severe and often less known, which lead to important comorbidities. With the aim of summarizing the available knowledge on neurosurgical intervention in patients with XLH, we present four emblematic cases with disorders requiring neurosurgical evaluation. Relevant diagnostic delay was seen in two of these cases, with another case demonstrating that complications requiring neurosurgery may be present even in patients with few symptoms. The last case stresses the need for care of adult patients who can present with undiagnosed comorbidities. We also carried out a narrative review on neurosurgical interventions in patients with XLH. Through case reports and a review of the literature, focus is placed on the role of the neurosurgeon in the management of patients with XLH. In fact, neurosurgical intervention can often provide stable outcomes for craniosynostosis and clinical improvement for symptoms related to spinal stenosis. Thus, the neurosurgeon can aid in optimizing management of patients with XLH and should be a member of both adult and pediatric multidisciplinary teams. Lastly, additional studies are needed to determine if the early use of burosumab in infants can help to avoid complications in the long term.
Keywords: Arnold-Chiari type 1 syndrome, Craniostenosis, Neurosurgical complications, X-linked hypophosphatemic rickets
Received: November 15, 2022
Accepted: February 13, 2023
Published online: March 7, 2023
AboutOpen - ISSN 2465-2628 - www.aboutscience.eu/aboutopen
© 2023 The Authors. This article is published by AboutScience and licensed under Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0). Commercial use is not permitted and is subject to Publisher’s permissions. Full information is available at www.aboutscience.eu
Introduction
X-linked hypophosphatemic rickets (XLH) is an X-linked dominant rare disease caused by mutations in the Phosphate-Regulating Endopeptidase X-Linked (PHEX) gene (Xp22.11). The protein encoded is a phosphate-regulating endopeptidase that regulates the expression of fibroblast growth factor 23 (FGF23) (1). Aberrations in the PHEX protein give rise to high serum concentrations of FGF23, which inhibit the activity of 1α-hydroxylase in the kidney, thus leading to hypophosphatemia and low levels of serum vitamin 1,25-dihydroxyvitamin D (2). Patients affected by XLH show marked hypophosphatemia, and decreased synthesis of 1,25-dihydroxyvitamin D, rickets, osteomalacia, odontomalacia, and short stature, in addition to complications including osteoarthritis, enthesopathies, spinal stenosis, and hearing loss (2,3). XLH is the most common form of inherited phosphate wasting with a prevalence that ranges from 1.7 per 100,000 children to 4.8 per 100,000 adults (4,5). Patients frequently develop clinical symptoms during the first or second year of life (2). Initial treatment consists of supplementation with oral phosphate and active vitamin D (2). However, in a substantial proportion of patients treatment is not entirely successful (6-8), especially in terms of lower limb deformities, short stature, and dental abscess. In addition, around 65% of children with XLH will require orthopedic surgery (6-9). This is probably related to the aim of treatment approaches toward reduction of alkaline phosphatase (ALP) serum levels to improve rickets and promote skeletal growth without normalization of serum phosphate (2).
Starting in 2018, to overcome unsuccessful management of XLH, a new treatment approach has been made available, namely burosumab, which is a fully human monoclonal IgG1 antibody that neutralizes FGF23, and approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) based on the results of pivotal clinical trials (10-12). In children with XLH, burosumab has been shown to improve renal tubular phosphate reabsorption, serum phosphate levels, growth, and rickets (10-12). In addition, burosumab had a good safety profile and is associated with clinically relevant benefits for patients in terms of pain and physical health scores (12,13).
Due to the rarity of XLH and the wide range of clinical manifestations, diagnosis is sometimes delayed, especially in mild cases. Moreover, management of the disease can be challenging due to several associated clinical implications that may arise sooner or later during follow-up. Therefore, it is widely accepted that the management of XLH patients requires a multidisciplinary approach (2). The neurological complications associated with XLH are the most severe and often less known, which lead to important comorbidities for patients. In most cases, when neurological symptoms arise neurosurgical evaluation and intervention is often required. Craniosynostosis and Arnold-Chiari type 1 malformation leading to syringomyelia in some cases are two of the most severe complications of XLH that should be considered during management. However, only a few reports are available in the literature and there is limited evidence about the management of patients with XLH and disorders requiring neurosurgery (14-16).
With the aim of summarizing the available knowledge on neurosurgical intervention in patients with XLH, we present four emblematic cases with disorders requiring neurosurgery. We also carried out a narrative review on neurosurgical interventions in patients with XLH. These aspects are worthy of discussion in light of the increasingly evident need for multidisciplinary management of patients with XLH.
Case presentations
Case 1
This female patient was born in 2000 and diagnosed with XLH at the age of 3.5 years, presenting with tibial varus, short stature, and anomalies of dental enamel. Molecular analysis confirmed the suspicion of XLH with the presence of a heterozygous frameshift mutation (c2019_2021delACCinsCCTCA [p.Pro674LeufsX14]) in the PHEX gene. The child started treatment with phosphate salts and calcitriol. During follow-up, improvement of rickets was achieved with regular stature growth, below the midparent target height. Body mass index (BMI) was always in the normal range for age and sex. Growth and biochemical parameters at diagnosis are shown in Table I.
At the age of 14 years, the patient experienced pain in the right forearm and reduced sensitivity in the right hand. Since the symptoms persisted, the patient underwent a neurologic evaluation that did not find cranial nerve deficits in strength, function, or coordination, and symmetrical reflexes were evoked at all four limbs. An electroneurographic study was performed with negative results. For persistent hypoesthesia in the arm and right hand, three years after the onset of the first symptoms, the patient underwent a cervical magnetic resonance imaging (MRI), which showed hypoplasia of the basiocciput and a small posterior cranial fossa, with descent of the cerebellar tonsils through the foramen magnum for a maximum distance of about 1 cm. A significant hydro-syringomyelic cavity was observed extending from the cranio-cervical region up to a plane passing through D10-D11, with a more evident finding in the mid-dorsal site (Fig. 1). The patient underwent occipito-cervical decompression surgery with coarctation of the cerebellar tonsils and autologous duroplasty with postoperative significant improvement of syringomyelia.
| Case 1 | Case 2 | Case 3 | Case 4 | |||||
|---|---|---|---|---|---|---|---|---|
| Diagnosis | Last visit | Diagnosis | Last visit | Diagnosis | Last visit | Diagnosis | Last visit | |
| Age (years) | 3 | 22 | 2 | 13 | 3 | 12 | 44 | 45 |
| Height (m) | 0.966 | 1.52 | 0.946 | 1.495 | 0.83
(–3.36) |
1.242
(–3.8) |
1.335 | 1.335 |
| Weight (kg) | 16.1 | 48 | 15 | 47 | 10
(–3.6) |
23.8
(–2.9) |
35.7 | 36.3 |
| BMI (kg/m2) | 17.3 | 21 | 16.8 | 21 | 14.5
(–0.56) |
15.4
(–0.4) |
20 | 20.5 |
| Pi (mg/dL) | 2.9 | 2.5 | 2.7 | 2.8 | 2.1 | 2.5 | 1.9 | 3.4 |
| ALP (IU/L) | 1413 | 143 | 1236 | 1096 | 908 | 290 | 67 | 59.9 |
| PTH (pg/mL) | 54 | 77 | 92 | 51 | 25 | 24 | 29.9 | 50.1 |
| 25-OHD (ng/mL) | N.A. | N.A. | N.A. | 15 | 9.8 | 8.5 | 18.7 | 13.2 |
| TRP (%) | N.A. | N.A. | N.A. | N.A. | 66.6 | N.A. | N.A. | 85.3 |
| TmP/GFR | N.A. | 2.13 | 1.67 | 2.32 | 1.40 | 2.9 | N.A. | 2.9 |
25-OHD = 25-hydroxyvitamin D; ALP = alkaline phosphatase; BMI = body mass index; GFR = glomerular filtration rate; N.A. = not available; Pi = inorganic phosphate; PTH = parathyroid hormone; TmP = tubular maximum reabsorption of phosphate; TRP = renal tubular reabsorption of phosphate.
See (26) as a reference for pediatric normal laboratory values.
In the years after surgery the patient did not show any severe neurological symptoms. There is some deformity at the level of the rib cage, with some pain, for she underwent orthopedic evaluation. In June 2021, she was started on burosumab (compassionate use, patient >20 years) with the aim of improving phosphate levels and painful symptoms. Growth and biochemical parameters at last visit are shown in Table I.
Case 2
A male patient is the second child of a mother affected by non-genetically confirmed XLH and not investigated for XLH prenatally or after birth. At 18 months of life, due to tibial varum the child underwent radiological evaluations at another center near his hometown. The X-ray survey showed no cranial bone lesions, thinning of the cranial theca, and moderate curvature of the femurs and tibias with varus knee and hypoplastic acetabular cavities. An enlarged and frayed appearance of the distal femoral and tibial metaphyses was seen, associated with bone trabeculation and slightly rarefied cortical profiles. Further biochemical evaluation showed elevated serum ALP (1,236 IU/L), reduced serum phosphate (2.7 mg/dL) with serum parathormone slightly above the upper normal level (92 pg/mL) (Tab. I), which confirmed the suspicion of hypophosphatemic rickets.
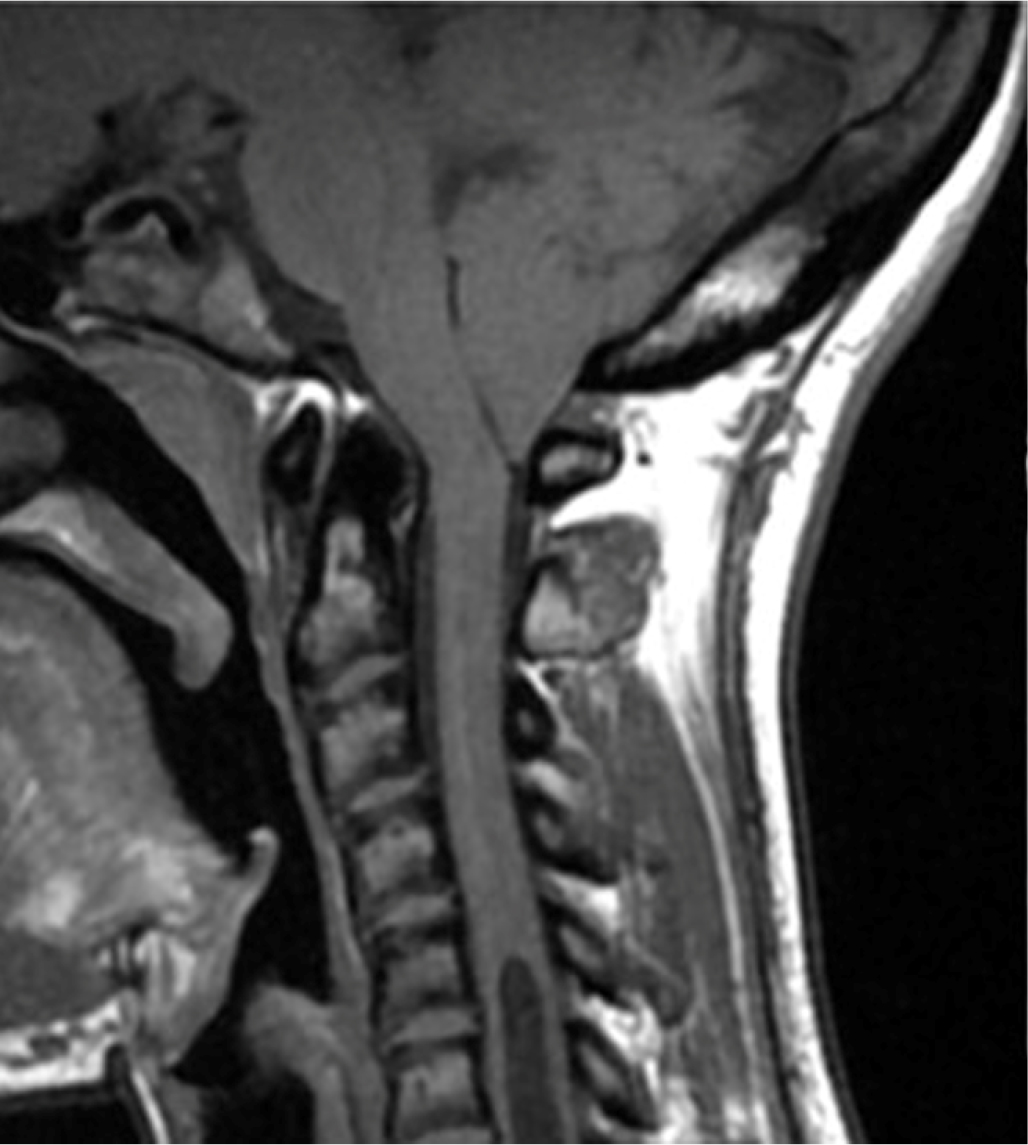
Fig. 1 - Magnetic resonance imaging showing significant hydro-syringomyelic cavity extending from the cranio-cervical region up to a plane passing through D10-D11 in case 1.
Molecular analysis of PHEX revealed the nonsense variation C.2104 C>T (p.R702X) in both the patient and his mother. The child promptly started treatment with phosphate salts and calcitriol. At 30 months, turricephaly, microcephaly, and prominence of the sagittal and coronal sutures were observed. Some episodes of headache associated with vomiting were reported and he was referred to our center. Computerized tomography (CT) showed pansutural craniosynostosis, enlarged fingerprints (Fig. 2), and ectasia of the cerebellar tonsils. Fundus oculi examination revealed papilledema and MRI of the brain showed tortuous optic nerves. The child was referred for neurosurgical evaluation.
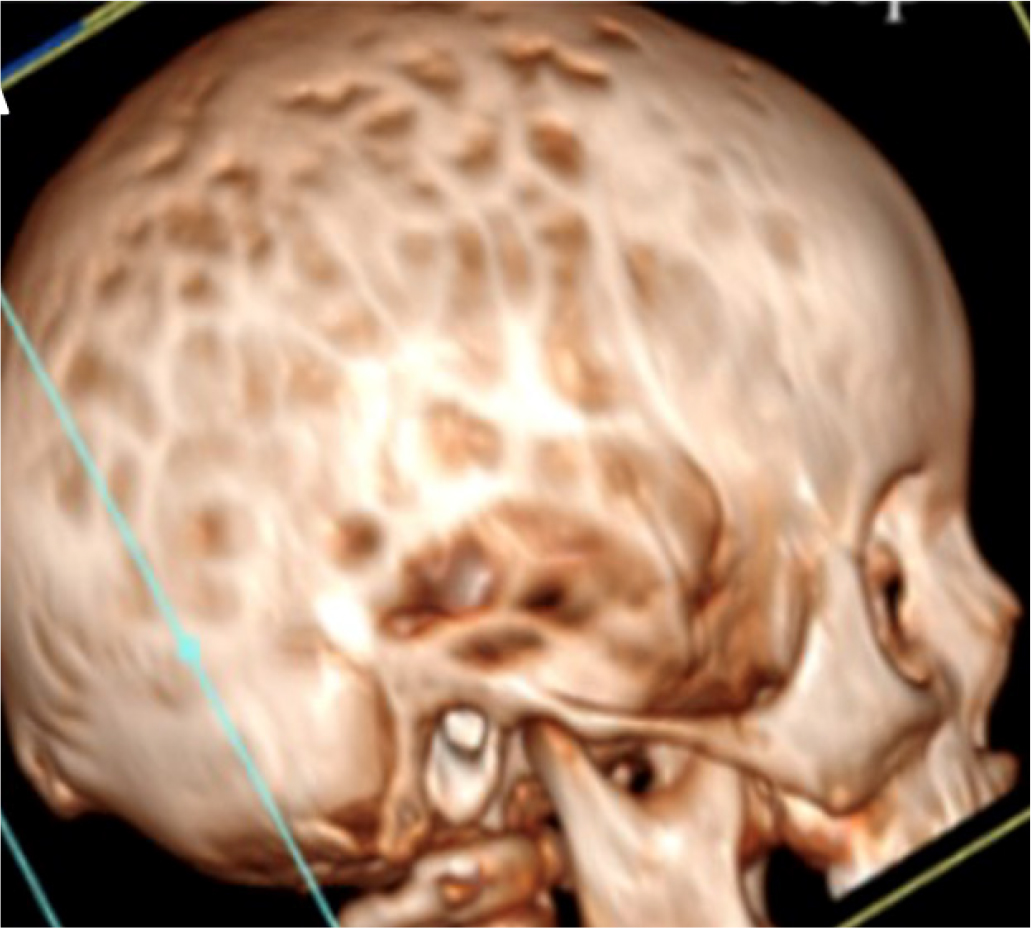
Fig. 2 - Cranial computed tomography image of case 2 showing pansutural craniosynostosis with enlarged fingerprints and ectasia of the cerebellar tonsils.
Invasive monitoring of intracranial pressure was initiated, confirming intracranial hypertension. The child underwent cranial vault expansion by distraction osteogenesis.
At postoperative follow-up, the patient showed dolichocephalic morphology and a stable brain MRI with no worsening of ectasia of the cerebellar tonsils (3 mm). Pale papilla was present on the right side, and the patient had reduced vision (right eye 4/10, left eye 8/10). Renal function and metabolic compensation were both within normal limits.
During follow-up the patient continued his regular treatment with active vitamin D and phosphate salts, regular monitoring of cranial MRI, renal ultrastructure, growth and skeletal features that eventually showed renal hyperechoic spots, lumbar scoliosis, plantar flatness, and minimal varus deformity of right tibia and femur. The growth trend was good and with straightening of the lower limbs. The patient did not show any dental abnormalities or abscess, and dental health was within normal limits. Growth and biochemical parameters at last visit are shown in Table I.
Case 3
A male patient with a family history of bone deformities and maternal short stature was diagnosed with XLH at 1.3 years of age for lower limb deformities and hypophosphatemia (PHEX mutation: p.Trp520Ter). Conventional therapy with alfacalcidol and phosphates was initiated at diagnosis with suboptimal results in both biochemical and growth responses.
At the age of 8 years, he started burosumab, with moderate biochemical response (improvement in ALP and Rickets Severity Score [RSS], but without recovery of hypophosphatemia; Tab. I). At the age of 11 years, the patient started to complain of headache and was referred for neurosurgical evaluation. Somatic characteristics related to rickets, with dolichocephalic caput, frontal bossing, and recess at the level of the coronal suture were observed. A high-resolution CT scan showed complete synostosis of the sagittal suture associated with marked reduction of the squamous suture.
A Chiari I malformation was diagnosed by MRI (Fig. 3A). No syringomyelia was seen at the level of the medulla. Venous sequences by MRI angiography showed the absence of signal at the level of the transverse sinuses, with the presence of collateral venous circulation (Fig. 3B, C), which could lead to intracranial hypertension due to reduction of liquor reabsorption in the venous system.
As the patient did not have papilledema, strict follow-up was started without neurosurgical intervention. Growth and biochemical parameters at last visit are shown in Table I.
Case 4
The patient is the 44-year-old mother of patient 3, and was previously misdiagnosed with achondroplasia. She was diagnosed with XLH at the age of 35 following diagnosis of her 11-month-old child. She had no family history of XLH. Stature and biochemical parameters at diagnosis are shown in Table I. At the age of 5, she underwent bilateral corrective tibial osteotomy for varus. Osteotomy for valgus deformity of the right knee was performed when she was 14 years old.
At initial evaluation, biochemical markers were calcium 9.0 mg/dL, phosphate 1.9 mg/dL, creatinine 0.59 mg/dL, ALP 67 U/L, parathyroid Hormone (PTH) 29.9 pg/mL, and 25-OH vitamin D 18.7 ng/mL. She was started on treatment with calcitriol 1 mg/day. Potassium dihydrogen phosphate plus disodium hydrogen phosphate (602 mg plus 360 mg), two tablets twice a day, was also added. In March 2022, she started treatment with burosumab (compassionate use, patient >20 years) to normalize phosphate levels and reduce pain. Biochemical parameters at last visit are shown in Table I. She referred that at the age of 43 she had undergone neurosurgical evaluation for headache.
An Arnold-Chiari type 1 malformation was diagnosed by MRI with cerebellar tonsils that engaged in the conjugation frame (for about 3 mm). Furthermore, the medullary cord at the level of the soma of C6 showed a modest extension of the ependymal canal of the syringomyelia type with clinically relevant cephalic expansion (Fig. 4). The patient chose to postpone the indicated surgical procedure.
Discussion
The present cases provide the opportunity to highlight several points regarding diagnosis and management of patients with XLH. In case 1, a severe Arnold-Chiari malformation was observed by MRI. Considering the first appearance of symptoms, imaging examination was not promptly carried out. We investigated the reasons of this diagnostic delay that could have been dramatically harmful for our patient. First, the patient mostly described her symptoms as a chronic condition with moderate pain or hypoesthesia in her hand. This condition may have been considered for treatment with anti-inflammatory drugs ex juvantibus, but not for immediate evaluation with cerebral MRI. However, we believe that the main reason for the diagnostic delay resides in the unawareness of Arnold-Chiari type 1 malformations in XLH patients, since this is the first patient we managed with this association. Notwithstanding, the patient had a good clinical outcome despite the severe radiologic picture. This may therefore raise the question of when MRI should ideally be performed to screen XLH patients for Arnold-Chiari type 1 malformation.
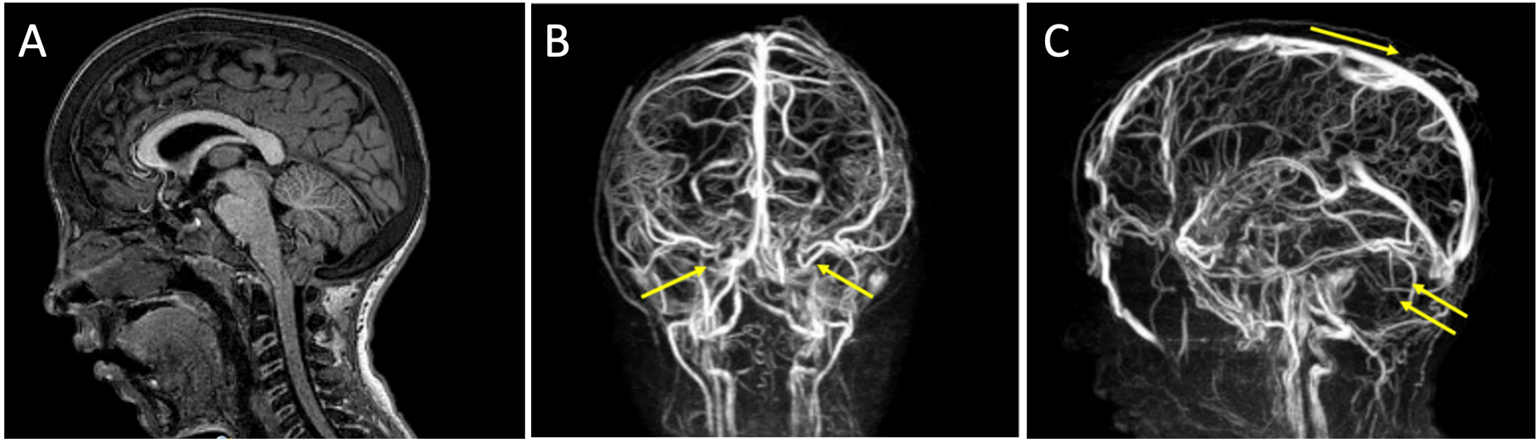
Fig. 3 - Case 3. A) Chiari type 1 malformation evident by MRI. B, C) Venous sequences by MRI angiography showing the absence of signal at the level of the transverse sinuses, with the presence of collateral venous circulation. MRI = magnetic resonance imaging.
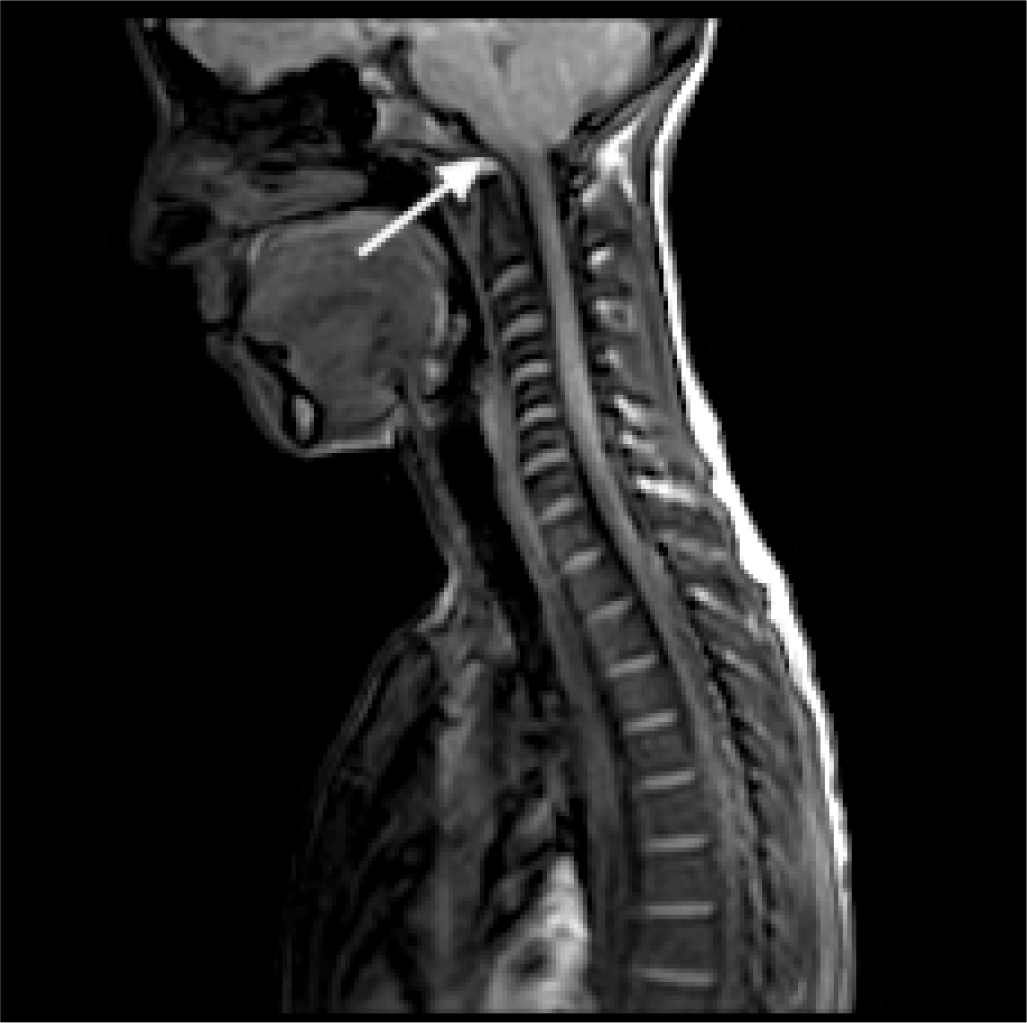
Fig. 4 - Magnetic resonance imaging of Arnold-Chiari type 1 malformation in case 4.
In case 2, a relevant diagnostic delay can also be noted, even if the patient was born to a mother known to have XLH but not genetically confirmed. It is possible to hypothesize that if the diagnosis of the mother had been genetically confirmed, her son would have been investigated in the first months of life by molecular analysis of PHEX. In our opinion, if the patient was correctly diagnosed with XLH due to PHEX variation in the first months of life, he could have undergone regular follow-up of skeletal features and neurosurgical workup, and thus surgery at an earlier age, which may potentially have avoided the patient’s symptoms. Craniosynostosis can be observed in about 60% of patients with XLH (14,15). Notwithstanding, only a fraction of patients will require surgery, which is possibly related to delayed or missed diagnosis of sutural synostosis. Therefore, all patients with XLH should undergo careful monitoring of cranial morphology and head circumference, and the patency of anterior fontanelle size during the first year of life (2). Other neurosurgical complications due to complete or partial fusion of cranial sutures include protrusion of the cerebellar tonsils developing the Arnold-Chiari malformation and in some cases syringomyelia (16).
Case 3 demonstrates that complications requiring neurosurgical evaluation may be present even in patients with few symptoms, and thus there is the need for a high degree of suspicion and early referral for proper follow-up. In addition, the question arose as whether to operate or not when faced with a rather late diagnosis of craniosynostosis in the absence of clear evidence of intracranial hypertension and no symptoms related to Chiari syndrome. Careful ophthalmologic, neurological, and neuroradiological monitoring is therefore mandatory in this case in order to evaluate the need for surgical treatment. Lastly, case 4 stresses the need for care of adult patients who can present with undiagnosed comorbidities. In this patient, XLH was diagnosed late only following a diagnosis of XLH in her child. Together, these cases demonstrate that clinicians should pay greater attention to complications of XLH, with an emphasis on early diagnosis and treatment. Clinical guidance is also needed regarding the possibility to carry out neurosurgical screening by nuclear magnetic resonance (NMR) in all patients diagnosed with XLH, or only in those with craniostenosis or its complications.
We carried out a literature search on PubMed to identify cases of XLH that were treated with neurosurgical approaches using the keywords: “X-linked hypophosphatemic rickets,” “neurosurgical complications,” “Arnold Chiari type 1 syndrome,” and “craniostenosis.” A total of 18 cases were identified (Tab. II). Broadly, two types of interventions were performed, cranial vault remodeling for craniosynostosis and laminectomy for spinal stenosis at various levels. These types of interventions are thus reflective of some of the most commonly observed associated complications of XLH. In all the cases reported, cranial vault remodeling was considered successful with a stable outcome at various follow-up times. In four of the five cases in which laminectomy was carried out, the outcome was considered favorable with clinical improvement. However, in the case reported by Kuether and Piatt in a 12-year-old patient with a large syrinx from C2 to T7 with significant foramen magnum stenosis and an Arnold-Chiari type 1 malformation, suboccipital craniectomy and C1 laminectomy with duroplasty did not lead to any significant improvement despite significant reduction in the diameter of the cervical thoracic spinal cord syrinx at 3 months after surgery (17). Two years after the intervention, the syrinx collapsed except for a small residual from T2 to T6.
In a cranial CT survey of 44 patients with XLH of variable age, even in the absence of symptoms, 26 cases of sagittal craniosynostosis were found, which was associated more frequently with the onset of dental abscesses and a higher incidence of Arnold-Chiari type 1 syndrome (15). The cranial index thus seems useful to identify patients in which it is easier to find an Arnold-Chiari type 1 syndrome. These authors highlight that sagittal craniosynostosis is frequent in patients with XLH, and that the association with Arnold-Chiari syndrome is also relatively common. Moreover, it was documented that cranial malformations appear to be associated with very severe rickets (15). In cases of craniosynostosis that occur after early childhood, hypophosphatemic rickets must be considered in the differential diagnosis (18).
In our case 1, the presence of an Arnold-Chiari type 1 malformation was not associated with craniosynostosis. Furthermore, the insurgence of neurological comorbidities in cases 1 and 2 does not seem to be related to the severity of rickets. Due to the rarity of disease, a clear correlation between genotype and phenotype in these two cases could not be evaluated.
| Author, year | No. of cases | Age at presentation | Reason for intervention | Intervention | Outcome |
|---|---|---|---|---|---|
| Vega et al., 2016 (18) | 7 | 1 to 9 years | Craniosynostosis, scaphocephaly | Cranial vault remodeling | Stable |
| Yamamoto and Onofrio 1994 (27) | 1 | 42 years | Severe spinal canal stenosis from T4 to T10 | Thoracic laminectomies | Favorable |
| Soehle and Casey, 2002 (28) | 1 | 44 years | Calcified intervertebral disc, marked cervical cord compression at C6/C7 | Anterior cervical discectomy/fusion with autologous bone graft | Favorable |
| Riccio et al., 2016 (29) | 1 | 49 years | Multilevel thoracic stenosis | Posterior decompression at T4-T5, T8-T9, T9-T10, and T11-T12, via a minimally invasive approach | Favorable |
| Kuether and Piatt, 1998 (17) | 1 | 12 years | Large syrinx from C2 to T7, with significant foramen magnum stenosis and a Chiari type 1 malformation | Suboccipital craniectomy and C1 laminectomy with duroplasty | No significant clinical improvement, after 2 years syrinx collapsed except for a small residual from T2 to T6 |
| Jaszczuk et al., 2016 (20) | 3 | >2 years | Sagittal suture craniosynostosis | Cranial vault expansion | Stable |
| Dunlop and Stirling, 1996 (30) | 1 | 49 years | Compression at T7, less at T5, T6, and T9 | Decompressive laminectomy from T6 to T10 | Full recovery |
| Murphy, 2009 (31) | 1 | 11 years | Craniosynostosis, scaphocephaly | Cranial vault expansion | Normalization |
| Migliarino et al., 2022 (32) | 2 | 7 years (twins) | Craniosynostosis, scaphocephaly | Cranial vault expansion | Not described |
In the literature, for many years these particular cranial conformations were not defined as craniosynostosis, but have often been associated with the underlying pathology, even if surgically treated scaphocephaly has been reported for decades. In 1984, it was hypothesized that cholecalciferol intoxication may trigger craniosynostosis (19). While today this seems like an unlikely hypothesis, there is still some concern that vitamin D may lead to premature closure of cranial sutures. In the 44-case cohort published by Rothenbuhler et al, in 60% of patients there was a synostosis of the sagittal suture and therefore scaphocephaly (15). While the shape of the skull is relatively unaltered, the suture of the skull is observed at later times, indicating the need for close observation, especially since clinical examination of the skull may provide evidence of cranial synostosis (20). In clinical practice, it is recommended to screen children with sagittal synostosis and children with Chiari malformation for hypophosphatemia and increased ALP, regardless of cranial morphology.
The presence of pain, physical abnormalities, and decreased function and mobility will further isolate patients with XLH with substantial psychological consequences (21). This is an example of the strong need, on the national level, for transition programs that include specific actions that patients view as priorities. Preparation for transfer and a multidisciplinary approach may thus enhance successful transition (22). The transition from childhood to adulthood is usually difficult in rare diseases. The number of patients that transfer to adult healthcare is still limited, with deep impact on their quality of life. It has been further suggested that specific pediatric and adult XLH teams be used to aid in the transition to adulthood care (23), and with attention to local protocols (24). This is also highly relevant considering that complete phenotypic rescue is rarely achieved in these patients, and chronic symptoms warrant multidisciplinary specialist care (25). Patient management and coordination of various medical disciplines is thus a cornerstone starting with diagnosis, and to optimize therapy and reduce the burden of disease (25).
With this in mind, herein the role of the neurosurgeon was highlighted, demonstrating that neurosurgical intervention can often provide stable outcomes for craniosynostosis and clinical improvement for symptoms related to spinal stenosis. Thus, the neurosurgeon can aid in optimizing management of patients with XLH and should be a member of both adult and pediatric multidisciplinary teams. Lastly, additional studies are needed to determine if the early use of burosumab in infants and adults can help to avoid complications in the long term.
Acknowledgment
This paper is dedicated to the memory of Dr. Filomena Carfagnini.
We would like to thank Patrick Moore who, on behalf of Health Publishing & Services Srl, provided publishing support and journal styling services.
Statement of Ethics
Written informed consent for publication (including images) has been obtained from the patients. The research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. The article is exempt from Ethical Committee approval since it is related only to four case reports.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Disclosures
Conflict of interest: FB reports to have participated in Advisory Boards for Kyowa Kirin, outside the submitted work. FA has nothing to disclose. FC has nothing to disclose. GLDG has nothing to disclose. DP reports to have received support for attending meetings and/or travel from Ipsen and to have held a leadership or fiduciary role in the Italian Society of Endocrinology, outside the submitted work. RS has nothing to disclose. GT has nothing to disclose. MZ reports to have received honorary for a lecture, outside the submitted work. AG reports to have been supported as speaker in SIEDP symposium in 2021 by Kyowa Kirin and to have participated in advisory board in 2020 and 2021 for the same company, outside the submitted work.
Financial support: Kyowa Kirin Srl funded the publishing support and journal styling services, besides an advisory board where paper concept was first discussed. Kyowa Kirin Srl had no role in the conduct of the literature search, in the preparation of the article, in the interpretation of clinical cases, and in the writing of the article for publication.
References
- 1. Imel EA, Econs MJ. Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol. 2005;16(9):2565-2575. CrossRef PubMed
- 2. Haffner D, Emma F, Eastwood DM, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435-455. CrossRef PubMed
- 3. Feng JQ, Clinkenbeard EL, Yuan B, White KE, Drezner MK. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone. 2013;54(2):213-221. CrossRef PubMed
- 4. Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. 2009;160(3):491-497. CrossRef PubMed
- 5. Endo I, Fukumoto S, Ozono K, et al. Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J. 2015;62(9):811-816. CrossRef PubMed
- 6. Kocaoglu M, Bilen FE, Sen C, Eralp L, Balci HI. Combined technique for the correction of lower-limb deformities resulting from metabolic bone disease. J Bone Joint Surg Br. 2011;93(1):52-56. CrossRef PubMed
- 7. Matsubara H, Tsuchiya H, Kabata T, Sakurakichi K, Watanabe K, Tomita K. Deformity correction for vitamin D-resistant hypophosphatemic rickets of adults. Arch Orthop Trauma Surg. 2008;128(10):1137-1143. CrossRef PubMed
- 8. Sharkey MS, Grunseich K, Carpenter TO. Contemporary medical and surgical management of X-linked hypophosphatemic rickets. J Am Acad Orthop Surg. 2015;23(7):433-442. CrossRef PubMed
- 9. De Cicco A, Toro G, Grandone A, et al. X-linked hypophosphatemic rickets. What the orthopedic surgeon needs to know. Int. J Bone Fragility Rare Common Disord. 2021;1(2):59-66. CrossRef
- 10. Carpenter TO, Imel EA, Ruppe MD, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(4):1587-1597. CrossRef PubMed
- 11. Carpenter TO, Whyte MP, Imel EA, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378(21):1987-1998. CrossRef PubMed
- 12. Insogna KL, Briot K, Imel EA, et al; AXLES 1 Investigators. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. J Bone Miner Res. 2018;33(8):1383-1393. CrossRef PubMed
- 13. Padidela R, Whyte MP, Glorieux FH, et al. Patient-reported outcomes from a randomized, active-controlled, open-label, phase 3 trial of burosumab versus conventional therapy in children with X-Linked hypophosphatemia. Calcif Tissue Int. 2021;108(5):622-633. CrossRef PubMed
- 14. Caldemeyer KS, Boaz JC, Wappner RS, Moran CC, Smith RR, Quets JP. Chiari I malformation: association with hypophosphatemic rickets and MR imaging appearance. Radiology. 1995;195(3):733-738. CrossRef PubMed
- 15. Rothenbuhler A, Fadel N, Debza Y, et al. High incidence of cranial synostosis and Chiari I malformation in children with X-linked hypophosphatemic rickets (XLHR). J Bone Miner Res. 2019;34(3):490-496. CrossRef PubMed
- 16. Watts L, Wordsworth P. Chiari malformation, syringomyelia and bulbar palsy in X linked hypophosphataemia. BMJ Case Rep. 2015;2015. CrossRef
- 17. Kuether TA, Piatt JH. Chiari malformation associated with vitamin D-resistant rickets: case report. Neurosurgery. 1998;42(5):1168-1171. CrossRef PubMed
- 18. Vega RA, Opalak C, Harshbarger RJ, et al. Hypophosphatemic rickets and craniosynostosis: a multicenter case series. J Neurosurg Pediatr. 2016;17(6):694-700. CrossRef PubMed
- 19. Carlsen NL, Krasilnikoff PA, Eiken M. Premature cranial synostosis in X-linked hypophosphatemic rickets: possible precipitation by 1-alpha-OH-cholecalciferol intoxication. Acta Paediatr Scand. 1984;73(1):149-154. CrossRef PubMed
- 20. Jaszczuk P, Rogers GF, Guzman R, Proctor MR. X-linked hypophosphatemic rickets and sagittal craniosynostosis: three patients requiring operative cranial expansion: case series and literature review. Childs Nerv Syst. 2016;32(5):887-891. CrossRef PubMed
- 21. Hamdy NAT, Harvengt P, Usardi A. X-linked hypophosphatemia: the medical expert’s challenges and the patient’s concerns on their journey with the disease. Arch Pediatr. 2021;28(7):612-618. CrossRef PubMed
- 22. Baroncelli GI, Mora S. X-linked hypophosphatemic rickets: multisystemic disorder in children requiring multidisciplinary management. Front Endocrinol (Lausanne). 2021;12:688309. CrossRef PubMed
- 23. Giannini S, Bianchi ML, Rendina D, Massoletti P, Lazzerini D, Brandi ML. Burden of disease and clinical targets in adult patients with X-linked hypophosphatemia. A comprehensive review. Osteoporos Int. 2021;32(10):1937-1949. CrossRef PubMed
- 24. Laurent MR, De Schepper J, Trouet D, et al. Consensus recommendations for the diagnosis and management of X-linked hypophosphatemia in Belgium. Front Endocrinol (Lausanne). 2021;12: 641543. CrossRef PubMed
- 25. Raimann A, Mindler GT, Kocijan R, et al. Multidisciplinary patient care in X-linked hypophosphatemic rickets: one challenge, many perspectives. Wien Med Wochenschr. 2020;170(5-6):116-123. CrossRef PubMed
- 26. Andropoulos BD. Gregory’s pediatric anesthesia. 5th ed. In: Gregory GA, Andropoulos DB, ed. Blackwell Publishing Ltd; 2012. Accessed October 14, 2022. Online
- 27. Yamamoto Y, Onofrio BM. Spinal canal stenosis with hypophosphatemic vitamin D-resistant rickets: case report. Neurosurgery. 1994;35(3):512-514. CrossRef PubMed
- 28. Soehle M, Casey AT. Cervical spinal cord compression attributable to a calcified intervertebral disc in a patient with X-linked hypophosphatemic rickets: case report and review of the literature. Neurosurgery. 2002;51(1):239-242. CrossRef PubMed
- 29. Riccio AR, Entezami P, Giuffrida A, Dowling J, Forrest G, German JW. Minimally invasive surgical management of thoracic ossification of the ligamentum flavum associated with X-linked hypophosphatemia. World Neurosurg. 2016;94:580.e5-580.e10. CrossRef PubMed
- 30. Dunlop DJ, Stirling AJ. Thoracic spinal cord compression caused by hypophosphataemic rickets: a case report and review of the world literature. Eur Spine J. 1996;5(4):272-274. CrossRef PubMed
- 31. Murthy AS. X-linked hypophosphatemic rickets and craniosynostosis. J Craniofac Surg. 2009;20(2):439-442. CrossRef PubMed
- 32. Migliarino V, Magnolato A, Barbi E. Twin girls with hypophosphataemic rickets and papilloedema. Arch Dis Child Educ Pract Ed. 2022;107(2):124-126. PubMed